染料敏化可见光催化制氢研究进展
2015-01-04李越湘彭绍琴南昌大学化学系南昌33003衡阳师范学院化学与材料科学系湖南衡阳4008
刘 兴 李越湘 彭绍琴 赖 华(南昌大学化学系,南昌33003;衡阳师范学院化学与材料科学系,湖南衡阳4008)
染料敏化可见光催化制氢研究进展
刘 兴1,2李越湘1,*彭绍琴1赖 华2
(1南昌大学化学系,南昌330031;2衡阳师范学院化学与材料科学系,湖南衡阳421008)
染料敏化是拓展宽禁带光催化剂激发波长范围、有效利用太阳光中可见光部分的重要手段.本文介绍了染料敏化分解水制氢的基本原理,综述了染料敏化剂、基质或载体、染料与基质的相互作用、产氢助催化剂以及电子牺牲剂的研究进展.并对光敏化体系中电荷转移途径及稳定性问题进行了讨论.
光催化制氢;染料敏化;敏化剂;敏化基质;稳定性
1 引言
随着全球能源危机的不断加剧,寻找新的绿色、高效能源已成为共识.H2被认为是最理想的替代能源载体之一.自1972年Fujishima和Honda1报道TiO2光电化学分解水产生H2和O2以后,掀起了半导体光催化制氢研究热潮.2-7但是由于TiO2(锐钛矿)的禁带宽度为3.2 eV,要在紫外光的激发下才有催化活性,而太阳光中紫外光仅占4%,加上光生电子-空穴极易快速复合及逆反应的发生,使得TiO2光催化制氢的总效率很低.染料光敏化是拓展TiO2等宽禁带半导体光催化剂激发波长范围、有效利用可见光的重要手段.8-12
染料敏化半导体的研究主要集中在Grätzel型染料敏化太阳能电池(DSSC),13而在光解水制氢方面相对较少.染料敏化光解水制氢是指,在可见光照射下,吸附于基质表面的染料分子吸收光子后被激发,激发态染料分子将电子注入到敏化基质(半导体)导带,最后在助催化剂作用下还原H+(H2O)而产生氢气(见图1).一个染料敏化光催化制氢体系一般由如下几个组成部分构成:(1)染料分子,(2)敏化基质(即电子接受/传递体),(3)产氢助催化剂,(4)电子牺牲剂.本文讨论的助催化剂为无机固体材料,不包含近些年来出现各种Ni、Co、Fe等配合物分子催化剂.14,15
©Editorial office ofActa Physico-Chimica Sinica
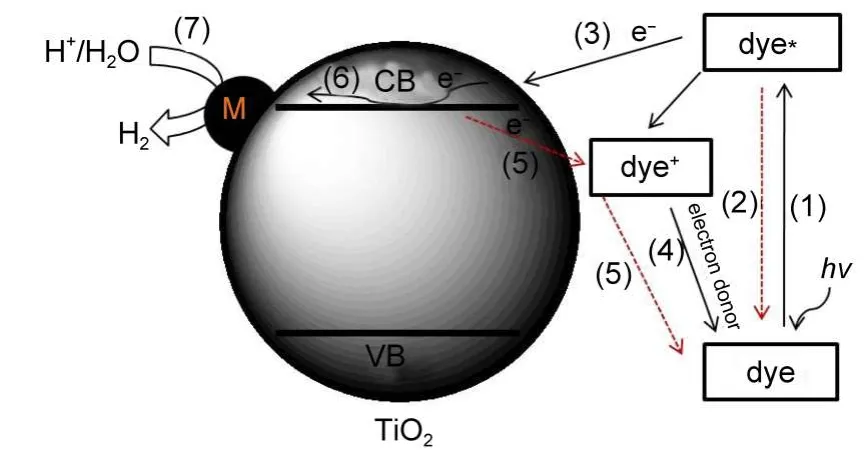
图1 染料敏化分解水制氢原理示意图Fig.1 Schematic illustration of dye-sensitized water splitting for hydrogen production
2 染料敏化分解水制氢基本原理
如图1所示,以TiO2基质为例,通常认为,染料敏化分解水制氢一般包括以下几个过程:16(1)基态染料分子(dye)受光激发产生激发态(dye*);(2)激发态dye*通过荧光等途径去激发而回到基态;(3)染料激发态将电子注入TiO2导带(CB);(4)失去电子产生的氧化态染料物种(dye+)通过与电子给体反应而获得再生;(5)注入半导体的电子回流到dye+而重新生成基态分子dye;(6)电子从半导体导带迁移到表面;(7)负载的助催化剂(如贵金属M)捕获电子而在其表面还原H2O(H+)产生H2.
由图1可知,对于染料敏化半导体,被光激发的是染料分子,而不是半导体,激发态染料将电子注入TiO2导带,促进了染料的电荷分离,这也相当于扩展半导体的光吸收范围.由步骤(1)、(3)、(4)、(6)和(7)构成了染料敏化制氢的完整过程.而染料敏化中有两个复合过程会降低染料的光利用率和电荷分离,即步骤(2)和(5)(分别是(1)和(3)的逆过程).根据以上分析,要实现有效的染料敏化制氢,染料敏化体系需要满足如下条件:8,10(a)染料能有效地吸收可见光产生激发态;(b)激发的染料高效地将电子注入半导体;(c)氢气在半导体表面的快速生成(存在高效的放氢助催化剂);(d)合适的再生剂(电子牺牲剂)使染料再生.
3 染料的类型与敏化方式
3.1 染料敏化剂的类型
根据染料敏化分解水制氢的原理,染料敏化剂作为“天线”分子,起着收集能量的作用,类似于自然界光合作用中叶绿素和胡萝卜素起的作用.因此,染料分子对敏化分解水制氢效果至关重要,一般地,作为敏化剂的染料应满足以下几个基本条件:17,18
(1)分子本身具有宽的光谱吸收范围,而且摩尔吸光系数大,这样才能有效地利用太阳光中的可见部分.
(2)其电子最低未占据轨道(LUMO)的能量应该高于半导体导带边缘的能量,有利于光激发染料的电子的转移;或半导体和染料能级有良好的轨道重叠,有利于能量的转递.
(3)光、热等稳定性好,且要求其激发态或氧化态在电子牺牲剂作用下再生能力好.
(4)能与半导体等基质材料表面较牢固、有效地结合,这要求染料分子中含有―COOH、―OH、―SO3H、―PO3H2等能与表面基团产生强的相互作用的官能团.
满足上述要求的敏化剂主要有金属有机配合物及不含金属的有机染料等.19
3.1.1 金属有机配合物染料
这类敏化剂主要有多吡啶钌染料、金属酞菁及金属卟啉等.多吡啶钌配合物由于具有稳定性相对较好、激发态寿命长等优点,成为研究得最多的敏化剂之一.这类染料按其结构可分为羧酸多吡啶钌、膦酸多吡啶钌、多核联吡啶钌等,其中羧酸、膦酸多吡啶钌的吸附基团分别为羧基、膦酸基.多吡啶钌配合物受光激发后主要发生金属-配体电荷转移(MLCT)跃迁(电子从钌的t2g轨道跃迁到配体的π*轨道).多吡啶钌配合物在DSSC研究中被广泛使用,13,20,21其用于光催化分解水制氢也有不少报道.22-29例如,早在1980年代初,Grätzel等22就报道了乙二胺四乙酸二钠盐(EDTA)作为牺牲剂存在下,联吡啶钌Ru(bpy)32+作为光捕获物质实现了可见光下分解水产氢;Bi和Tien23也报道了Ru(bpy)32+-TEOA (三乙醇胺)-Rh(bpy)33+-Pt体系可以在可见光下较高效地进行光催化分解水得到氢.Dhanalakshmi等24对[Ru(dcbpy)2(dpq)]2+(dcbpy:4,4'-二羧基-2,2'-联吡啶;dpq:2,3'-双-(2-吡啶基)-喹喔啉)敏化的TiO2催化剂在可见光下光催化产氢进行了研究,发现最佳条件下产氢速率可达0.28 mL·h-1.Zhang等25研究了不同双核联吡啶染料[Ru2(bpy)4(BL)](ClO4)2、[Ru2(dcbpy)4(BL)](ClO4)2、Ru(dcbpy)2(NCS)2(BL:桥连配体)敏化未负载贵金属的TiO2光催化制氢,发现第一种染料的敏化效率最高,对应体系在27 h的染料转换数达75340,420和475 nm单色光的表观量子效率分别为16.8%与7.3%,作者把这种差别归咎于它们跟TiO2之间的配位形式、电子注入、电子回流等的性质不同所致.最近,Veikko等30合成了如图2(a)所示的双核钌敏化剂Ru2(bpy)4BL(ClO4)2,其具有良好共轭性、大的分子面积、与基质TiO2较好结合等优点,相比Ru(bpy)2L(ClO4)2(图2(b)),该敏化剂显示了更高的敏化可见光(λ>400 nm)分解水制氢活性与稳定性,其激发后同时产生MLCT和金属-金属电荷转移(MMCT),而对这两种电荷转移方式之间的协同效应机理,作者未作深入的探讨.
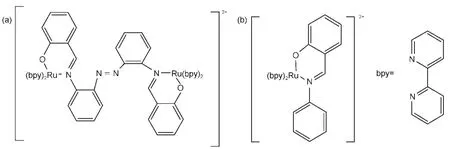
图2 (a)Ru2(bpy)4BL(ClO4)2与(b)Ru(bpy)2L(ClO4)2分子结构30Fig.2 Molecular structures of(a)Ru2(bpy)4BL(ClO4)2and(b)Ru(bpy)2L(ClO4)230
金属酞菁(酞菁结构如图3(a))是大π共轭配合物,酞菁类化合物可以看成是异吲哚的衍生物,它由四个异吲哚环组成封闭的十六元环.酞菁类化合物有两个吸收带,一个在大约在550-700 nm处中等吸收强度的Q带,另一个在大约350-400 nm处为B带(也称Soret带).酞菁具有储备电子与输出电子的双重功能,既可作为电子给体又可以作为电子受体.Oman等31用酞菁修饰Fe-TiO2与TiO2(P25),在长波范围产生光吸收明显提高(获得)了可见光催化放氢活性.吉仁17以酞菁铜四磺酸四钠为敏化剂敏化了Pt/TiO2,以KI为电子牺牲剂实现了可见光分解水制氢,但产氢速率很低(仅1.4 μmol·h-1),如何提高酞菁类敏化剂的产氢速率有待进一步研究.
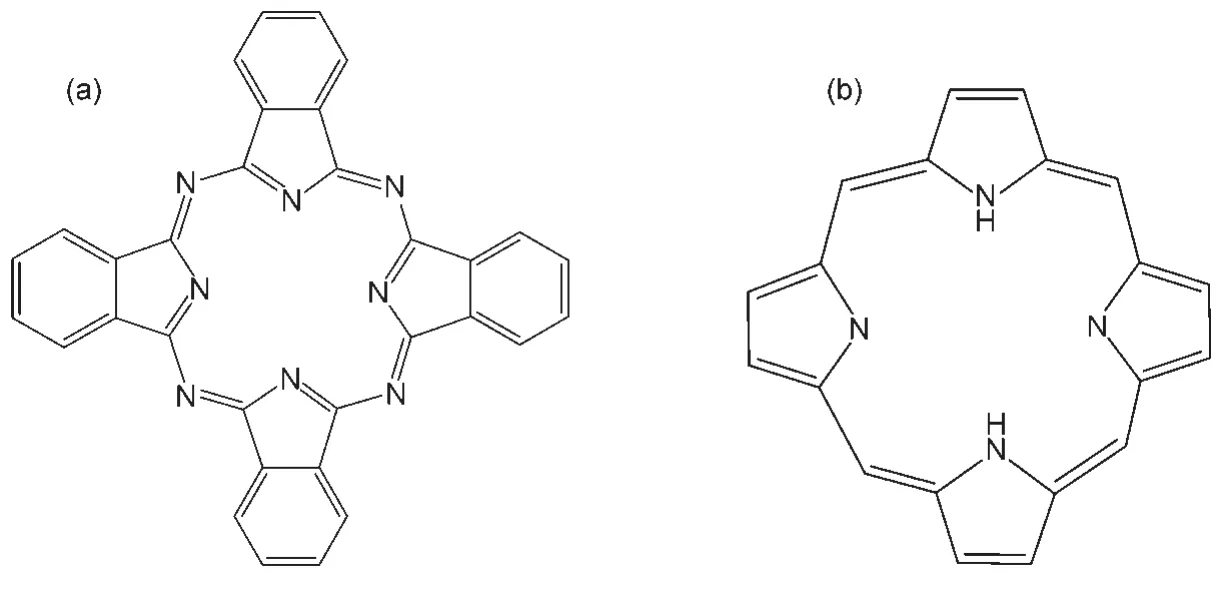
图3 酞菁(a)与卟啉(b)分子结构Fig.3 Molecular structures of phthalocyanine(a)and porphyrin(b)
卟啉(如图3(b))是自然界广泛存在的含四聚吡咯的杂环化合物,能与铁、镁、锌等离子相结合,形成含四个N原子的平面结构.这类化合物作为敏化剂用来制氢已有一些报道.如,Harriman等321981年就研究了锌卟啉水溶液中还原水产氢情况. Hagiwara等33报道了利用铬卟啉(Cr-TPP)敏化一系列硫化物(SnS、CdS、ZnS、NbS2、Ln2S3、Cr2S3、Ag2S),在可见光下,SnS、Ag2S被敏化后光催化活性均有显著提高(Na2S-K2SO3为电子给体),作者还指出Cr-TPP的Soret带吸收对敏化效果起了主要作用.Kim等34用锡卟啉(SnP)敏化TiO2,在pH 3-11的范围均可以实现分解水产氢,作者认为自由基阴离子SnP-·是光敏化制氢的中介物种,其有足够长的寿命,可以从溶液体相扩散到TiO2表面再注入电子,因此染料不需要吸附在基质表面,然而高浓度的SnP-·容易发生使卟啉环共轭性破坏的不可逆氢化,因此该体系制氢的稳定性有待解决.
除此之外,还有一些其它的金属有机配合物也可以用作光解水制氢的敏化剂,如Astuti等35研究了锌取代的细胞色素c(ZnCyt-c)对TiO2、SnO2等氧化物的敏化作用,发现ZnCyt-c激发三重态可以将电子高效注入氧化物电极,产生长寿命(0.4 s)的电荷分离态TiO2(e-)/ZnCyt-c+,使得该体系获得了(10± 5)%的分解水产氢量子效率.
3.1.2 不含金属的有机染料
不含金属的有机染料种类繁多,成本较低,吸光系数高,而且便于进行分子设计,近来引起不少研究者的兴趣.占吨类染料是一类研究得最多的有机染料(结构见图4),主要包括曙红(EY)、荧光素(FL)、罗丹明B(Rh B)、虎红(RB)、藻红(ER)、福禄考红(Ph)等衍生物.从分子结构看出,占吨类染料分子中含有氧化蒽环,且有可解离的羧基和酚羟基,是一类二元酸,随pH值增加,其结构会呈现内酯、醌式、单阴离子、两性离子、双阴离子等不同形式,36因此这类染料的电子吸收光谱、荧光光谱等强烈依赖于溶液的pH值,一般最大吸收波长在490-560 nm之间,这部分波长的光在太阳光谱中正好是相对较多的.
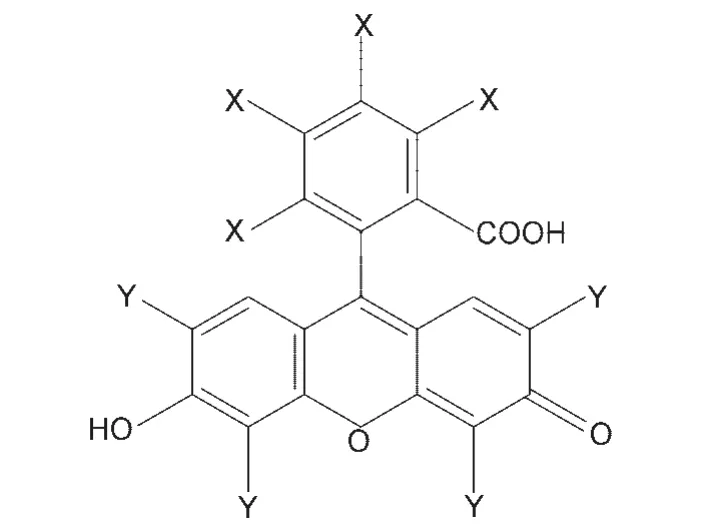
图4 几种常见占吨染料的结构36Fig.4 Structures of several common xanthene dyes36
当浓度较高时,占吨染料容易在溶液中发生自聚集,其中尤以二聚的情况比较普遍,37,38染料的聚集现象可以简单地通过其紫外-可见吸收光谱来判断.以EY为例,在水溶液中的浓度为10-6mol·L-1,其最大吸收波长在515-520 nm左右,且在490 nm有一肩峰,而当浓度达到10-4mol·L-1,此两峰的强度基本相当,490 nm的吸收峰对应于EY的H型(面对面)二聚体.此外,EY还有J型(肩并肩)二聚形式.38,39这两种二聚体形式在其它占吨染料中也同样存在.38染料的聚集状态对敏化制氢活性有较大的影响,如聚集体中的分子间能量转移会导致激发态电子得不到利用而使敏化制氢活性降低.40二聚体也可在光激发后产生正、负离子自由基对,如EY≡EY二聚体激发得到EY-·…EY+·,41分离后的EY-·可将多余的电子转移给基质(助催化剂)从而达到敏化分解水制氢的目的.42,43
占吨染料还有一个重要性质就是它们的取代基效应(重原子效应).一些含重原子(如卤素)的占吨染料除具有很高的荧光量子产率外,还容易发生隙间窜跃,产生长寿命的三重激发态,44这对激发态将电子注入电子受体是极为有利的.41通常,含重原子的占吨染料的敏化制氢活性高,Shimidzu等45分析了这一现象,并研究了外加卤素离子产生的外部重原子效应对占吨染料敏化制氢活性影响.荧光素是一个具有很强荧光发射的物质,其不同基团取代后的衍生物荧光性质有所改变,占吨染料跟其它物质相互作用后,也可以通过荧光光谱的变化来判断电荷转移特征,46这对于研究染料敏化机理是一个有力的工具.
由于占吨染料的这些优点,其用于光敏化分解水制氢具有较早的历史.1985年Mau等47研究了甲基紫精(Xanthene-MV2+)-乙二胺四乙酸(EDTA)-Pt光解水制氢,并指出其中的染料相当于光合作用中的能量天线分子;Heleg与Willner48也报道了可见光下EY修饰的Pd-TiO2制取氢气.Gurunathan等49用不同的染料敏化Pt-RuO/SnO2体系,发现产氢速率由大到小依次为:曙红>玫瑰红>联吡啶>罗丹明B≈吖啶黄>荧光素;Abe等50同样使用不同占吨染料敏化Pt/TiO2,以三乙醇胺作电子牺牲剂重点考察了它们分解水制氢的活性高低,其中也是以曙红的活性最高.
EY(含溴取代原子)是占吨染料中作为敏化剂研究得最多的一种之一,其敏化制氢活性高.如本课题组51研究了EY敏化不同温度制备的TiO2,随着TiO2锻烧温度的降低,比表面积与表面羟基密度增加,染料EY的吸附量也不断增加,因此制氢活性相应提高.Chatterjee等52研究了包括EY、罗丹明B等在内的染料激发态不同氧化还原电位对敏化制氢活性的影响.Sreethawong等53研究了EY敏化自制的介孔TiO2及不同的商品TiO2,比较发现EY敏化负载Pt的介孔TiO2比负载Pt的商品TiO2电荷传输效率更高,具有更高的产氢活性.
除占吨类染料外,还有一些不含金属的有机染料被报道可应用于光敏化分解水放氢,如花菁类、香豆素、苝染料等.Abe等54发现部花青和香豆素敏化的Pt/TiO2在乙腈的水溶液中可见光下可以制氢.刘福生等55报道了苝染料N,N'-二(4-吡啶基)-3,4,9, 10-苝四羧酸二酰亚胺敏化Pt/TiO2光催化可见光分解水制氢.Maia等56研究了甲基橙、亚甲基蓝、铬黑T等染料对β-BiTaO4的敏化作用,其中β-BiTaO4/亚甲基蓝体系在可见光下经历诱导期后显示了分解水制氢活性.Zhang和Choi57发现酚醛树脂与TiO2可形成表面复合物,在可见光下两者之间存在配体-金属电荷转移(LMCT),从而获得了可见光制氢活性.
染料敏化太阳电池中有机染料分子有种比较普遍的设计理念,即电子供体-π-电子受体(D-π-A)结构,58-60鉴于DSSC与染料敏化光催化制氢原理上的类似性,近年来有学者也尝试将前者的D-π-A有机染料引入到染料敏化光解水制氢中,如Peng等61用吲哚基D-π-A染料LI-4(图5)敏化负载Pt的石墨状氮化碳聚合物C3N4(g-C3N4)在可见/近红外光区(λ≥ 420 nm)得到了233.8 μmol·h-1的制氢活性,而LI-4与锌酞菁衍生物Zn-tri-PcNc(图5)共敏化负载Pt的g-C3N4的制氢活性则达371.4 μmol·h-1,两种染料的协同效应明显(见3.2节敏化方式).
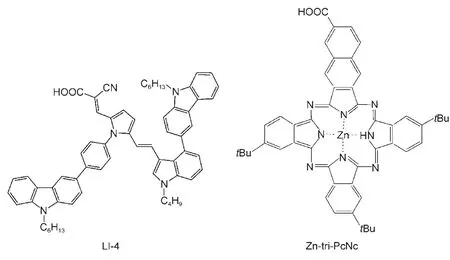
图5 D-π-A有机染料LI-4与锌酞菁衍生物Zn-tri-PcNc的分子结构61Fig.5 Molecule structures of D-π-Aorganic dye LI-4 and zinc phthalocyanine derivative Zn-tri-PcNc61
3.2 敏化方式
3.2.1 单一染料敏化
按照体系中所用敏化剂数目的多少,可以把敏化方式分为单一染料敏化与多种染料共敏化.在前面我们讨论的染料敏化多为单一染料敏化制氢体系.
3.2.2 多种染料共敏化
由于单一染料吸收光谱一般只限于某一波长范围,不能充分利用可见光,多种染料共敏化能进一步扩展光吸收范围,是实现高效利用太阳光的途径之一.近些年来,随着越来越多的染料被用作光敏剂,染料的协同敏化效应越来越多地被报道.如, Zhao等62设计合成了系列方酸菁染料,它们的吸收光谱与钌配合物有很好的互补性,在600-700 nm左右呈现出一个很强的吸收带,吸光系数较N3 (Ru(II)(dcbpy)2(SCN)2)染料高一个数量级,取得了较好的协同敏化效果.Min和Lu63用EY和RB两种占顿染料共同敏化石墨烯/Pt等材料,实验发现这两种染料具有协同敏化效果,在520和550 nm两个波长的可见光同时照射下,量子效率可高达37.3%,而且这种协同敏化作用对TiO2/Pt及多壁碳纳米管/Pt同样存在;进一步研究认为,64EY与RB对TiO2的共敏化作用不仅与染料对可见光的吸收增加有关,而且还与染料分子之间的荧光共振能量转移有关,后者可有效降低染料激发态由于荧光猝灭导致的能量损失,因而提高了光催化产氢效率.
4 敏化基质与载体
4.1 敏化基质
用于敏化的基质材料作为染料光生电子的接受体,热力学上要求半导体的导带位置应当比敏化剂的激发态或还原态能级低(更正).如TiO2(锐钛矿)的导带电位为-0.50 V(pH 7.0,vs NHE(标准氢电极),下同),65而联吡啶钌Ru(bpy)23−激发态电位为-0.84 V,66EY的激发态电位为-1.1 V,42因此, Ru(bpy)23−与EY激发态均可将电子注入TiO2导带.除了TiO265,67,68外,还有很多其它材料可以接受染料光生电子而用作基质材料,如SnO2,69钙钛矿钛酸盐(MgTiO3、CaTiO3、SrTiO3),70K4Nb6O17纳米卷,66Ti-MCM-41分子筛,71多层碳纳米管(MWCNT),72Na2Ti2O4(OH)2,73多金属氧酸盐,74还原石墨烯,75,76碳氮聚合物C3N4,77-79凹凸棒80等.Zhang等81用EY敏化TS-1分子筛,获得了9.4%的表观量子效率,作者推测TS-1分子筛表面的“―O―Ti4+―O―”链接受了染料的光生电子,进而在Pt作用下产氢;而在染料敏化Ti-MCM-41分子筛中,同样是“―O―Ti (IV)―O―”转移了光生电子.71MWCNT作为转移电荷的载体,能够捕获其吸附的染料分子的激发态电子再传递至Pt位点而还原H2O(H+)制氢,有效阻止了电子-空穴复合,从而获得了12.14%的可见光(λ≥420 nm)量子效率.72Min和Lu75,77研究了EY敏化载铂的还原态石墨烯(RGO)及多孔石墨状C3N4(mpg-C3N4)可见光制氢情况,得出RGO优异的电子接受能力和转移电荷能力,使得其能够有效地转移染料光生电子到催化剂Pt,从而获得了较高的产氢量子效率;而mpg-C3N4作为一种新型优良的不含金属的共轭碳氮化合物,同样可以有效促进染料光生电子的转移,550 nm可以得到19.4%的量子效率.本课题组78比较了不同温度下制备的g-C3N4对EY敏化制氢的影响,发现低缺陷的g-C3N4的染料吸附量大,敏化制氢活性高;类似地,提高RGO的导电性,有利于染料电荷转移,从而提高敏化制氢活性.76
近来,金属-有机骨架(MOF)材料作为一种新型有机-无机杂化纳米多孔材料,也被用作敏化基质,82-84如Mori及其合作者82报道了Ru(bpy)23+敏化MOF[Ru2(p-BDC)2]n(BDC:对苯二羧酸盐),在EDTA作为电子牺牲剂及甲基紫精(MV2+)作为电子传递剂下,实现了可见光照下产氢,相关反应过程如图6所示.

图6 [Ru2(p-BDC)2]n,Ru(bpy)23+,MV2+,EDTA混合体系可见光照下光催化产氢机理82Fig.6 Mechanism of photocatalytic hydrogen evolution in the mixture system of the [Ru2(p-BDC)2]n,Ru(bpy)23+,MV2+,and EDTAunder visible light irradiation82
4.2 基质的修饰与改性
为提高染料敏化的效率,对敏化基质的表面修饰与改性是一种有效的方法.Kim等85报道覆盖Al2O3薄层的TiO2作为基质,可以阻止注入导带的电子与敏化剂Ru(bpy)23+氧化态之间的复合,从而显著提高了光解水活性.Jin等86制备了EY敏化的CuO (1.0%(w))/TiO2催化剂,TiO2担载的CuO能与EY以多齿的配位作用改善TiO2对染料的吸附性能,从而提高光敏化制氢活性,使表观光量子效率提高到5.1%,CuO还有利于电荷分离、电子转移、改善水还原状态等,在可见光下,催化剂在120 h内显示出良好稳定性.本课题组87发现,对TiO2进行氮掺杂,然后用EY敏化,发现氮的掺入显著地提高了可见光光催化分解水制氢的活性,究其原因是由于表面N掺杂产生的氧空位增强了染料分子在TiO2上的吸附.本课题组88,89还发现,由于EY有三个远离的配体(羧基、酚氧基和羰基),而金属离子,如Fe3+离子可以和EY分子进行配位作用,通过Fe3+这种偶联作用,在TiO2表面形成单层或具有三维网状聚合物结构多层的染料(如图7),增强了染料的吸附和催化剂对可见光的吸收,改善了电子的传输效率,从而大幅提高了光解水制氢的活性,λ>420 nm的可见光表观光量子效率达19.1%.
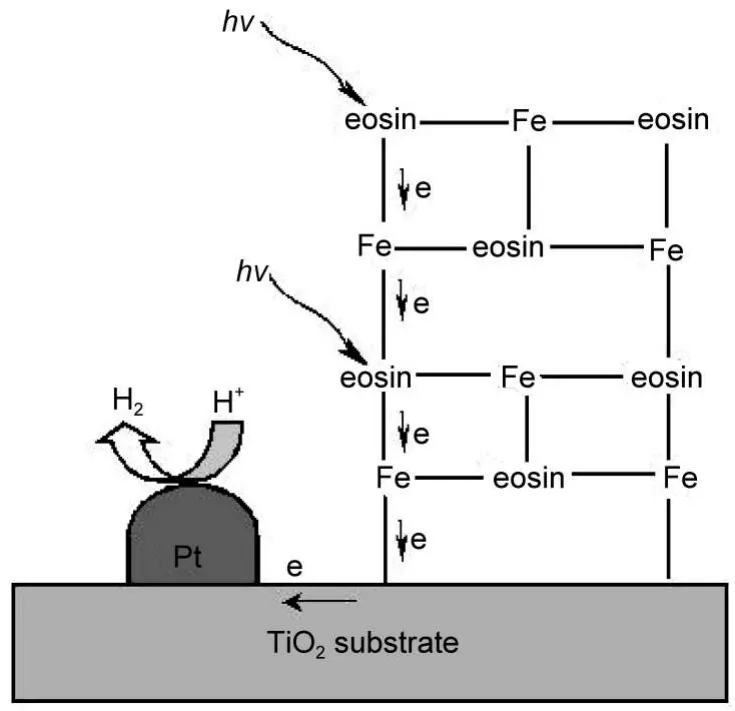
图7 经Fe3+连接的吸附多层EY的TiO2光催化制氢示意图89Fig.7 Schematic illustrations of multilayer adsorption of EY via linkage of Fe3+on TiO2for photocatalytic hydrogen evolution89
最近,我们65通过SO42−与PO43−修饰TiO2,也提高了其光敏化产氢活性.SO42−和PO43−能够通过化学作用与TiO2结合,其吸电子诱导作用可促进光生染料物种EY•-H(染料阴离子自由基的质子化形式)向催化剂导带的电子注入(如图8所示),而且修饰后的TiO2导带边位置发生负移,提高了导带上的电子还原水的能力.
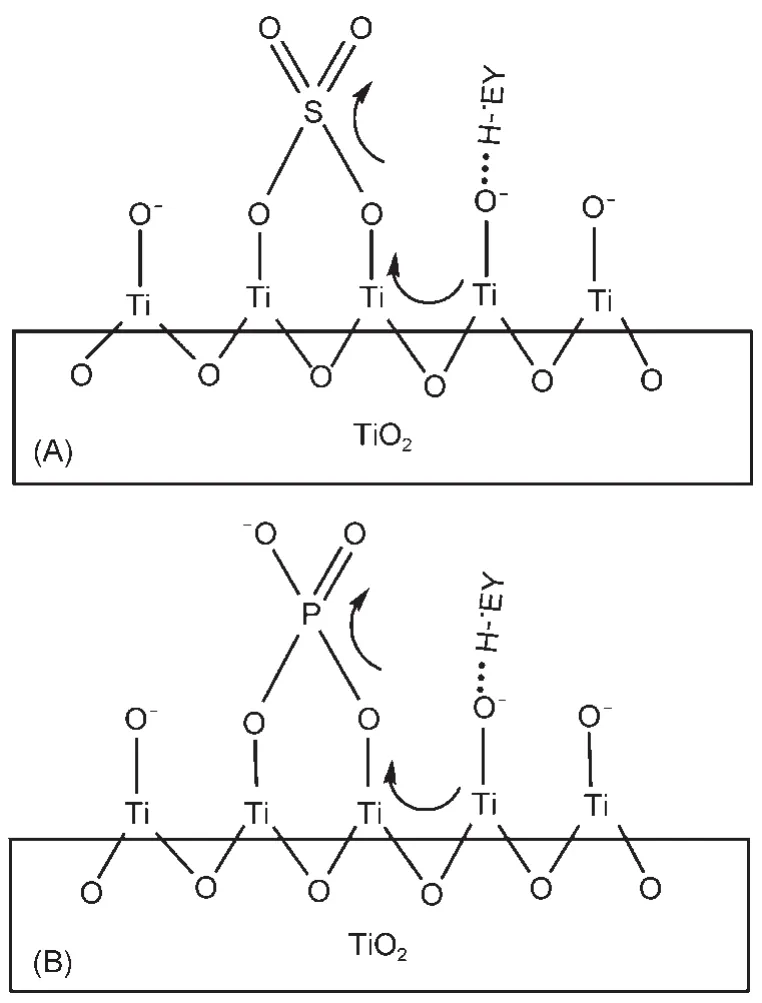
图8 键合在TiO2上的硫酸根(A)或磷酸根(B)的诱导效应及EY•-H与>Ti―O−之间的氢键的示意图65Fig.8 Schematic illustration of the inducing effect of the sulfate(A)or phosphate(B)bound at TiO2and the hydrogen bond between EY•-H and>Ti―O−65
4.3 载 体
光敏化中也有用到载体的,即材料本身一般并不接受光生电子,只是起到分离电荷作用.如Lu等分别采用硅胶(SiO2)81,90和坡缕石((Mg,Al)4Si8(O,OH, H2O)24·nH2O)42作为载体,以EY作敏化剂,在含三乙醇胺作电子给体的水溶液中显示了良好的可见光制氢活性,前者的放氢速率和量子效率分别可以达到43 μmol·h-1和10.4%,后者的放氢速率为3247.2 μmol·h-1,量子效率为12.5%,由于SiO2、Al2O3、MgO等的导带电位较高,染料不能将电子注入其中,而硅胶、坡缕石的表面荷负电,通过静电作用促进了EY二聚体(EO-EO)光激发后产生的正、负离子自由基(EO-·、EO+·)的分离,产生的EO-·可将电子转移至Pt位点,从而提高了制氢活性(如图9所示).
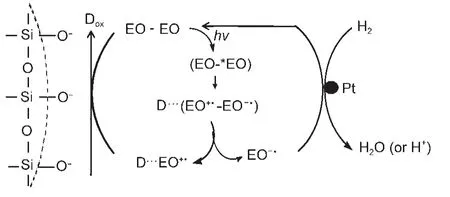
图9 EY/硅胶/Pt光敏化制氢的可能机理90Fig.9 Probable mechanism of photosensitized hydrogen evolution over EY/silica gel/Pt90
5 染料与敏化基质的相互作用
染料分子向半导体导带注入电子时,注入速率Kinj可表示为:91

式中:V是染料与半导体表面键合强度,h为Planck常数,ρ是电子态密度.由(1)式可知,染料分子与半导体的表面结合作用对电子注入速率起决定性作用.作为敏化剂的染料,分子中一般含有―COO-(或―COOH)、―SO3H、―PO3H2、―OH、―Si―O―、―HN―(C=O)―等锚定基团,与TiO2或其它基质的表面基团(原子)容易发生化学键作用和氢键作用.此外,静电作用也是染料与基质之间的常见作用力.
化学键作用力较强,比染料分子单纯仅依靠范德华力、氢键吸附到基质上更有利于电子的注入,染料与基质之间形成的常见化学键类型如图10所示.如Murakoshi等92通过红外(IR)光谱、紫外-可见(UV-Vis)光谱等手段表征羧酸多吡啶钌染料与TiO2表面的化学作用,报道了类酯键吸附模型(图10(a)).含有―COO−(或―COOH)的占吨染料与半导体的结合也有被认为通过(类)酯键与基质结合的.71,81Abe等93通过硅烷耦联剂将染料以化学方法固定在载铂TiO2表面从而构造一个稳定的染料敏化光催化体系,染料的转换数超过1×104,在520 nm处的光量子效率大约为10%,究其原因,作者认为是EY与硅烷耦联剂处理后的TiO2形成了在水中较稳定的化学键,显著提高了稳定性,但活性却较EY直接敏化的TiO2降低了很多.甲硅烷基因其对TiO2表面羟基的良好亲和性及形成的Si―O键的化学惰性,是理想的染料锚定基团(图10(d)).94Perera等69以AlCl3作为偶联剂,用RB敏化剂通过与SnO2形成“―(C=O)―O―Al(Cl)―O―Sn≡”键,在表面形成作用较强的吸附层,取得较高的光吸收和转换效率.Chen等95研究发现,含羧基、羟基、磺酸基等不同锚定基团的苯并噻唑类半花菁染料在TiO2表面的结合强弱及电子注入存在这样的次序:羧基+羟基>羧基>磺酸基+羟基,且认为同时含有羧基与羟基的半花菁染料与TiO2形成表面配合物,因此敏化效果最好.也有一些含双酚羟基的有机化合物,如儿茶酚或水杨酸与TiO2反应也能生成新的配位化合物而结合在TiO2表面.96Ikeda等97报道了联萘酚修饰的TiO2在三乙醇胺水溶液中的制氢.如图11,作者认为联萘酚的酚羟基与TiO2的表面羟基可以发生酯化或螯合作用而结合形成表面化合物(这两种结合均发生分子间脱水),但是没有给出相关的有力证据.
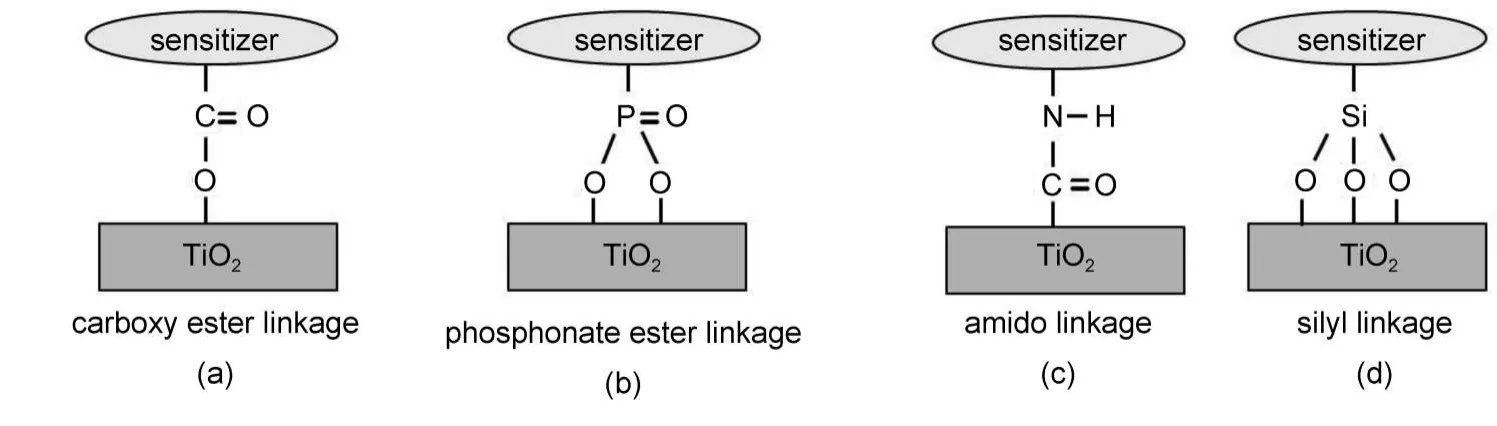
图10 一些含不同锚定基团的染料敏化剂与TiO2基质的键合方式19,92,98Fig.10 Some binding modes between dye sensitizers with different anchoring groups and TiO2matrix19,92,98
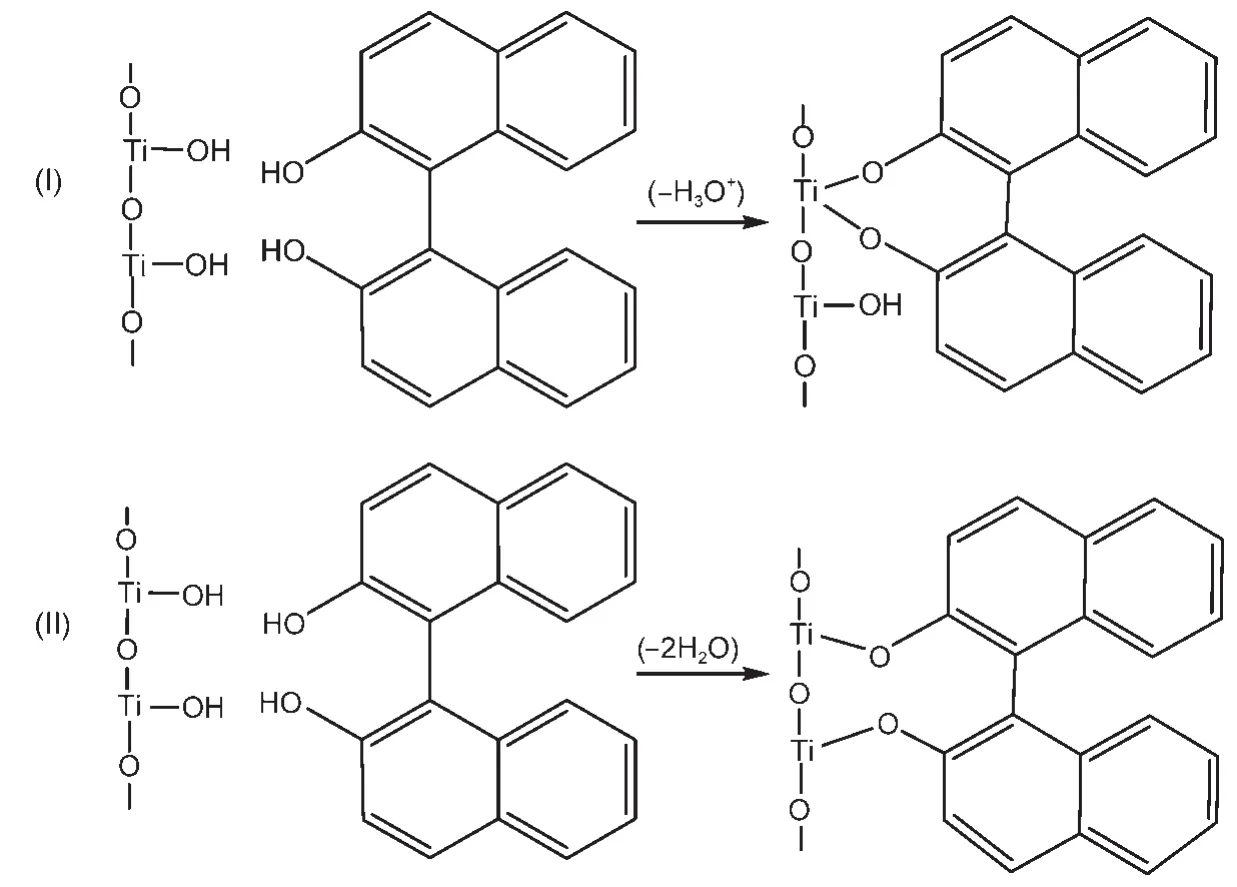
图11 联萘酚与Ti―OH之间通过(I)螯合(1:2加合),(II)酯化(2:2加合)的反应示意图97Fig.11 Schematic illustration of the reactions between Ti―OH and binaphthol through(I)chelation(1:2 adduct)and (II)esterification(2:2 adduct)97
Peng等99考察了三种吡啶钌配合物与TiO2表面的结合情况与制氢活性.其中双核吡啶钌配合物(Ru2(bpy)4L1-PF6)比单核吡啶钌配合物(n-Bu4N)2-cis-Ru(dcbpy)2(SCN)2(N719)和Ru(bpy)2(him)2-NO3的放氢活性好,作者认为双核配合物能把两个中心钌离子吸收的电子转移至TiO2表面,因此制氢活性高. Bae等100给出了几种联吡啶-钌配合物与TiO2表面结合的几种可能的方式(如图12所示),联吡啶-钌配合物中的―COOH可以与TiO2通过单齿或双齿配位形成表面化合物;―PO3H2则可以有单齿、双齿或三齿三种形式,通过漫反射红外光谱观察到―COOH的C=O、CO2-等键及―PO3H2的P=O、P―O―H、O―P―O等键的伸缩振动吸收峰的变化来判断结合的具体方式.他们29还系统地研究了联吡啶-钌配合物不同锚定基团对制氢活性影响,得出含膦酸基官能团的吸附能力强于羧酸官能团的染料,并且前者不易受到竞争吸附物(EDTA)的影响,因此获得了更高的敏化产氢活性.

图12 联吡啶-钌配合物(通过―COOH(a)或―PO3H2(b))与TiO2的可能结合形式100Fig.12 Possible combination modes between rutheniumbipyridyl complexes(through―COOH(a) or―PO3H2(b))and TiO2100
6 产氢助催化剂
光敏化制氢中通常会用到助催化剂,一般由贵金属单质(铂、钯、钌、铑等)充当,这是因为贵金属可以有效降低析氢的过电位,从而显著提高光催化分解水制氢活性.此外,贵金属的功函通常高于半导体(Pt有最高的功函),在半导体和贵金属之间形成Schottky能垒,促进了光生电荷分离.Jin等101研究了负载不同贵金属(铂、钌、铑)对EY敏化的TiO2可见光制氢活性的影响,发现通过贵金属负载显著地提高光量子效率,其中以负载铂效果最佳,最高光量子效率为10.27%.Li等26发现以铂的联吡啶配合物Pt(dcbpy)Cl2(dcbpy=4,4'-dicarboxy-2,2'-bipyridine)为铂源,通过光降解以化学配位作用锚定在TiO2上的该配合物而产生单质Pt,相比以H2PtCl6为铂源光沉积得到的Pt,前者具有更小的粒子尺寸与更高的分散性,从而有利于光敏化过程的电子在半导体和助催化剂间传递,因此达到了更好的光敏化制氢效果.
由于贵金属成本高,资源短缺,从实际应用的角度来说是不利的,需要寻找价廉、地球资源丰富的替代品.近年来,一些非贵金属的化合物被陆续报道可以用作光敏化制氢的助催化剂,如MoS2,79,102CoSx,103Ni(OH)2,40CuO/NiO,104CuO/CoO,105CuO/ Cr2O3106等,文献15综述了地球上丰富的物种作为光催化制氢助催化剂的研究进展情况.
一些光敏化制氢体系只有助催化剂而没有基质材料,或说基质材料和助催化剂是同一物质,如Zhang和Xu107研究了占吨染料敏化Ni、NiO、NiS、NiSe等系列镍基无机材料,其中以Erythrosin Yellowish敏化NiS活性最高,作者还比较了NiS不同制备方法对敏化制氢性能的影响,在聚乙二醇中制备的非晶态NiS具有不饱和Ni中心,能更充分与敏化剂接触,因此显示了最好的制氢性能.Lu等108考察了无电子转移中介物质存在下的Pt-Rose Bengal-TEOA体系可见光制氢的机理,认为染料二聚体在光激发、电荷分离与转移中起了关键作用.
7 染料的电荷转移、再生及稳定性问题
7.1 染料激发态的电荷转移
在多数染料敏化光解水制氢报道中,染料分子受激发后产生的激发态将LUMO轨道的电子注入半导体的导带,然后转移到助催化剂还原水产氢.而失去电子后的染料的氧化产物与电子牺牲剂反应得以再生,如图1和(2)-(6)式所示.这里,S、S+、S―表示敏化剂及其氧化态、还原态物种,1S*、3S*分别是敏化剂的单重、三重激发态,D、Dox是电子牺牲剂及其氧化产物,Sc表示半导体(基质).就激发态染料淬灭步骤来说,(4)是一个氧化淬灭途径.激发态染料淬灭还有还原途径((7)式).45而(6)和(9)式为染料淬灭后的两种再生方式.一般而言,在染料敏化体系中会同时存在以上(A)、(B)两种染料的电荷转移和再生途径.
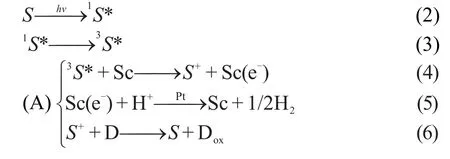

7.2 染料再生与电子牺牲剂
尽管染料存在还原和氧化两种再生方式,电子牺牲剂(或称电子给体)是染料再生过程中所必需的.电子牺牲剂一般由容易失去电子、具有较强还原性的物质充当.按照热力学,其氧化还原电位应比激发态或氧化态染料的电位更负.比较常见的电子牺牲剂有I−/I3−、17,18,22,54EDTA、12,18,29,35三乙胺、27二乙醇胺、71三乙醇胺,26,47,86-90甲醇28,109等.
金属有机配合物作为敏化剂的情况下,比较普遍使用的电子牺牲剂是EDTA及I−/I3−.如Abe等110采用部花青染料敏化的Pt/TiO2,在含乙腈和I−的溶液中实现了可见光分解水制氢,得到的量子效率为2%,研究证明,I−还原染料分子后自身变成I3−.该课题组54还发现,随乙腈含量减小,I−/I3−的氧化还原电位与染料分子HOMO能级之间差值变小,导致产氢速率降低.而在占吨类染料作为敏化剂的体系中,使用得最多的电子牺牲剂是三乙醇胺,如前面介绍的EY敏化的各种体系中绝大多数都使用了三乙醇胺作为电子牺牲剂.文献111研究了三乙醇胺在光催化反应过程中的作用机制,发现其分子中N原子上未共用孤电子对容易失去,是反应的活性位点.有关电子牺牲剂的选择规律及其与敏化剂性质之间的关系,目前还鲜有报道,特别是廉价、可再生的牺牲剂有待研究.
7.3 染料稳定性问题
在光敏化电荷转移过程中,激发态染料的还原淬灭产生(途径B)的S-自由基是一个不稳定的物种,容易发生单分子降解.这种降解对于一些含重原子(卤素)的占吨染料而言,S-发生光化学脱卤素现象严重.45,112-114而对一些金属配合物为敏化剂的体系, S-则容易发生配体脱除等反应,115-118如Probst等116分析发现[Re(CO)3(bipy)2]染料激发态被电子牺牲剂TEOA淬灭产生的还原产物易发生吡啶配体的脱除而降解.显然,敏化剂的降解会直接导致产氢活性下降甚至中止,使得体系使用寿命较短.为了避免或减少这种现象的发生,提高染料敏化体系的稳定性,一方面可以加强途径(A),或抑制途径(B),以减少生成S-的浓度;另一方面也可以在反应体系中加入高效稳定的电子受体(中继体)迅速转移S―的电子(中继体再将电子转移给基质或助催化剂),使其来不及发生降解就已经复原为基态染料,从而增强染料的稳定性.作为强化染料激发态氧化淬灭途径的一个例子,Eisenberg等112以FL为敏化剂,[Ni(pyS)3]-(1-,pyS=pyridine-2-thiolate)为催化剂构建了一个相对稳定的光敏化制氢体系(如图13),通过荧光光谱等证明,当电子牺牲剂三乙胺(TEA)的浓度较高(>0.36 mol·L-1)时,敏化剂激发态(1FL*)主要发生还原淬灭,产生不稳定的FL-自由基阴离子,其快速发生单分子降解,使得体系制氢活性显著降低;而当TEA的浓度较低(<0.36 mol·L-1)时,催化剂1-对1FL*的氧化淬灭远大于TEA的还原淬灭,此时生成的FL-浓度很低,因此敏化剂得以稳定,制氢体系虽然活性降低但是稳定性显著提高,当然这种稳定是动力学上相对的.
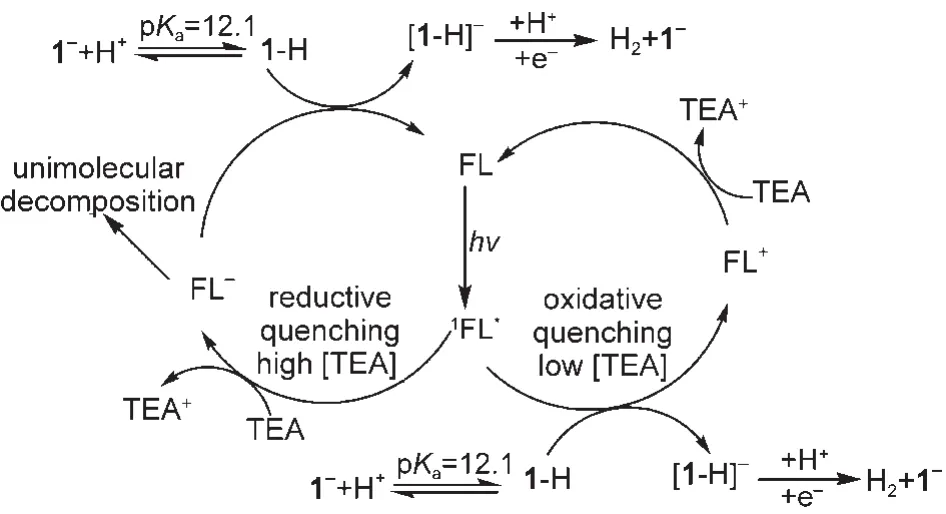
图13 FL敏化[Ni(pyS)3]-(1-)光致产氢反应机理113Fig.13 Reaction scheme for photogeneration H2in FL sensitized[Ni(pyS)3]-(1-)system113
李波和吕功煊119通过在EY敏化Pt/TiO2光催化体系中引入电子传递剂MV2+成功构建了高活性与稳定性的可见光催化产氢体系.MV2+可使EY激发态发生氧化淬灭并且转移还原淬灭产物EY−•的电子,有效降低了不稳定中间体EY−•的形成和积累,促进了电子由染料分子向产氢活性位点的有效传递,从而提高了产氢体系的活性和稳定性.
Hong等120采用自组装方法让Erythrosin B和Pt紧密有序地固载在层状双金属氢氧化物上,有利于还原淬灭激发态染料产生的自由基阴离子的电子传输到Pt,一定程度上抑制染料的脱卤素行为,因此这种“光敏剂/催化剂/载体”体系相对Erythrosin B-Pt体系制氢稳定性有所改善,但其染料降解仍十分明显.
我们74发现,多金属氧酸盐(POM)作为优良的电子受体,能够迅速转移光生电子,可以有效阻止染料分子的降解,从而构建了高效、高稳定性的染料敏化制氢体系.如图14,EY-α-[AlSiW11(H2O)O39]5--Pt体系(TEOA为电子给体)在可见光下显示了较高的分解水制氢活性与稳定性,五个光催化反应周期内,体系均维持活性不变,表观量子效率为10.3% (λ>420 nm),其中520 nm单色光下的量子效率达到28.0%.研究发现,pH=10.0的TEOA水溶液中,EY与AlSiW11具有化学键作用,这有利于它们之间的高效、迅速的电荷转移,有效抑制了染料的脱溴降解反应,大大提高了染料的光化学稳定性.
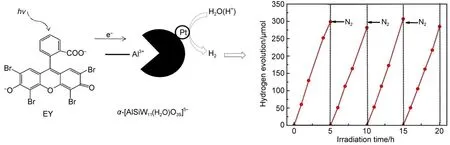
图14 EY-α-[AlSiW11(H2O)O39]5−-Pt体系光催化制氢示意图74Fig.14 Schematic illustrations of photocatalytic hydrogen evolution by EY-α-[AlSiW11(H2O)O39]5−-Pt system74
最近,我们121还用缺位的POM SiW11O8-39(SiW11)修饰TiO2,当SiW11通过化学键结合至TiO2表面后,由于SiW11具有强的接受电子能力,染料EY激发态被电子牺牲剂TEOA还原淬灭产生的EY−•(其质子化的形式EY•-H与SiW11或TiO2的表面氧原子可形成氢键)向SiW11LUMO轨道的电子转移十分高效:
EY•-H…(O)SiW11/TiO2➝EY+SiW-11/TiO2(10)形成的SiW-11又将电子快速注入TiO2的导带,

最后,电子被TiO2上的Pt捕获,还原H+而产生氢.

图15 EY敏化SiW11O83-9(SiW11)/TiO2产氢的反应机理121Fig.15 Reaction mechanism of hydrogen production in EY sensitized SiW11O83-9(SiW11)/TiO2121

图16 光照不同时间后EY-TiO2(A)和EY-SiW11/TiO2(B)反应液的吸收光谱121Fig.16 Absorption spectra of reaction solutions from EY-TiO2(A),EY-SiW11/TiO2(B)after different irradiation time121

以上过程如图15所示,SiW11作为一个良好的电子传递剂,极大地促进了EY•-H向TiO2导带的电子转移,从而有效抑制了EY-•的脱溴降解反应(图16(B),EY吸收光谱无明显变化),因此该体系具有较高活性与很好的稳定性,在λ>420 nm可见光下,20 h得到了11.4%的平均表观量子效率.而无SiW11的EY-TiO2的体系(图16(A)),吸收光谱蓝移, EY降解显著.
8 展望
目前为止,已在光敏化制氢方面做了不少的工作主要围绕以下几个方面进行的:(a)新型染料分子(生色团)的设计;(b)基质材料(或载体)的筛选与合成;(c)染料与基质材料的结合方式及表面结构探讨;(d)非贵金属光敏化制氢体系研究.总体来讲,尽管已取得一定成果,但仍然面临着不少问题,如,由于染料分子本身容易发生氧化还原反应而被降解等因素导致的敏化剂和体系的稳定性差,工作寿命不长等问题,使得染料敏化分解水制氢目前仅局限于基础研究阶段.要解决这些问题,进一步提高染料敏化制氢的效率与稳定性,一方面在于敏化基质,目前所报导的光敏化半导体制氢体系所采用的敏化基质大多为表面积较小的材料,如二氧化钛等.需要开发结晶度好、多孔的、表面积大的敏化基质,如前文提过的一些MOF新型材料有很好的晶体结构,规则的孔道,较高的稳定性,可在分子水平上设计和调控,合成方法多样等优点,122是一种不错的选择.此外还有特殊晶面占优的半导体(TiO2)123-125等也是比较有潜力的基质材料.另一方面是设计、合成吸收波长范围宽、吸光强度大且能级合适的染料敏化剂.借鉴DSSC对有机染料的研究成果,将D-π-A结构的染料用作光敏化制氢的敏化剂是一个有前景的方向.126-128这种结构有利于电荷分离和电子向基质的传输,同时扩大分子吸收波长(红移),拓展捕获光子的范围,且可以通过各个部分的设计控制调节染料的LUMO/HOMO能级、激发态寿命、氧化态/还原态的稳定性等,这对于光敏化制氢是至关重要的.此外,在染料敏化分解水制氢中需要加入电子牺牲剂,很多作为电子牺牲剂的物质并不常见且成本不低,从实际应用的角度,这是不利的,如果不使用电子牺牲剂就需要同时存在一个水氧化的催化剂,有报道三氧化钨(WO3)是水氧化产氧的良好催化剂.126
我们认为今后工作的重点包括:
(1)对现有染料进行改性,以及多种染料的共敏化,从而进一步匹配太阳光谱.
(2)合成新型廉价稳定的D-π-A有机染料作为敏化剂,增大其吸光效率.
(3)高效、非贵金属的制氢助催化剂的开发,特别是分子型助催化剂.
(4)染料敏化光催化机理的深入探讨.
(5)构建高效、稳定的光敏化体系,包括可再生的牺牲剂或无牺牲剂的反应体系.
(6)对新型基质材料的修饰及控制合成.
(1) Fujishima,A.;Honda,K.Nature 1972,37,238.
(2) Kudo,A.;Miseki,Y.Chem.Soc.Rev.2009,38,253.doi: 10.1039/b800489g
(3) Chen,X.B.;Shen,S.H.;Guo,L.J.;Mao,S.S.Chem.Rev. 2010,110,6503.doi:10.1021/cr1001645
(4) Huang,Y.F.;Wu,J.H.Prog.Chem.2006,18(7-8),168. [黄昀方,吴季怀.化学进展,2006,18(7-8),168.]
(5) Maeda,K.;Teramura,K.;Lu,D.L.;Takata,T.;Saito,N.; Inoue,Y.;Domen,K.Nature 2006,440,295.doi:10.1038/ 440295a
(6) Osterloh,F.E.Chem.Mater.2008,20,35.doi:10.1021/ cm7024203
(7) Li,Y.X.;Lü,G.X.;Li,S.B.J.Mol.Catal.(China)2001,15 (1),72.[李越湘,吕功煊,李树本.分子催化,2001,15(1), 72.]
(8) Ni,M.;Leung,M.K.H.;Leung,D.Y.C.Chin.J.Power Sources 2006,30(10),856.[倪 萌,Leung,M.K.H., Leung,D.Y.C.电源技术,2006,30(10),856.]
(9) Zhang,X.J.;Li,S.B.;Lü,G.X.J.Mol.Catal.(China)2012, 24(6),569.[张晓杰,李树本,吕功煊.分子催化,2012,24 (6),569.]
(10) Pei,D.H.;Luan,J.F.Int.J.Photoenergy 2012,2012,86.
(11) Ni,M.;Leung,M.K.H.;Leung,D.Y.C.;Sumathy,L.K. Renew.Sust.Energy Rev.2007,11,401.doi:10.1016/j. rser.2005.01.009
(12) Justinyoungblood,W.;Anna,L.S.;Maeda,K.;Mallouk,T.E. Accounts Chem.Res.2009,42,1966.doi:10.1021/ar9002398
(13) O'Regan,B.;Grätzel,M.Nature 1991,353,737.doi:10.1038/ 353737a0
(14) Du,P.W.;Eisenberg,R.Energy Environ.Sci.2012,5,6012. doi:10.1039/c2ee03250c
(15) Ran,J.R.;Zhang,J.;Yu,J.G.;Jaroniec,M.;Qiao,S.Z.Chem. Soc.Rev.2014,43,7787.doi:10.1039/C3CS60425J
(16) Pan,G.F.Modification of TiO2,ZnO and Their Performanceof Photocatalytic Hydrogen Evolution under Visible Light. Masteral Dissertation,Nanchang University,Nanchang,2007. [潘高峰.TiO2、ZnO的改性及其可见光催化制氢性能[D].南昌:南昌大学,2007.]
(17) Ji,R.Study on Hydrogen Production from Water Photolysis Using Phthalocyanine Dye Sensitized Nano TiO2.Nanjing Agricultural University,Nanjing,2007.[吉 仁.酞菁染料敏化纳米TiO2光解水制氢的研究[D].南京:南京农业大学, 2007.]
(18) Liang,M.;Tao,Z.L.;Chen,J.Chem.Online 2005,No.12, 889.[梁 茂,陶占良,陈 军.化学通报,2005,No.12, 889.]
(19) Fresno,F.;Hernández-Alonso,M.D.Green Energy Technol. 2013,329.
(20) Nazeeruddin,M.K.;Kay,A.;Rodicio,I.;Humphry-Baker,R.; Mueller,E.;Liska,P.;Vlachopoulos,N.;Grätzel,M.J.Am. Chem.Soc.1993,115,6382.doi:10.1021/ja00067a063
(21) Nazeeruddin,M.K.;Pechy,P.;Renouard,T.;Zakeeruddin,S. M.;Humphry-Baker,R.;Comte,P.;Liska,P.;Cevey,L.; Costa,E.;Shklover,V.;Spiccia,L.;Deacon,G.B.;Bignozzi, C.A.;Grätzel,M.J.Am.Chem.Soc.2001,123,1613.doi: 10.1021/ja003299u
(22) Grätzel,M.Accounts Chem.Res.1981,14,376.doi:10.1021/ ar00072a003
(23) Bi,Z.C.;Tien,H.T.Int.J.Hydrog.Energy 1984,9,717.
(24) Dhanalakshmi,K.B.;Latha,S.;Anandan,S.;Maruthamuthu, P.Int.J.Hydrog.Energy 2001,26,669.
(25) Zhang,X.H.;Veikko,U.;Mao,J.;Cai,P.;Peng,T.Y.Chem. Eur.J.2012,18,12103.doi:10.1002/chem.201200725
(26) Li,J.;E,Y.;Lian,L.S.;Ma,W.H.Int.J.Hydrog.Energy 2013,38,10746.doi:10.1016/j.ijhydene.2013.02.121
(27) Kruth,A.;Hansen,S.;Beweries,T.;Bruser,V.;Weltmann,K. D.ChemSusChem 2013,6,152.doi:10.1002/cssc.201200408
(28) Peng,T.Y.;Dai,K.;Yi,H.B.;Ke,D.N.;Cai,P.;Zan,L. Chem.Phys.Lett.2008,460,216.doi:10.1016/j. cplett.2008.06.001
(29) Bae,E.;Choi,W.J.Phys.Chem.B 2006,110,14792.doi: 10.1021/jp062540+
(30) Veikko,U.;Zhang,X.B.;Peng,T.Y.;Cai,P.;Cheng,G.Z. Spectrochim.Acta A 2013,105,539.doi:10.1016/j. saa.2012.12.061
(31) Oman,E.S.;Navio,J.;Litter,M.J.Adv.Oxid.Tech.1998,3 (3),261.
(32) Harriman,A.;Porter,G.;Marie-Claude,R.J.Chem.Soc., Faraday Trans.2 1981,77(5),833.doi:10.1039/ f29817700833
(33) Hagiwara,H.;Matsumoto,H.;Ishihara,T.Electrochemistry 2008,76(2),125.
(34) Kim,W.;Tachikawa,T.;Majima,T.;Li,C.;Kim,H.;Choi,W. Energy Environ.Sci.2010,3,1789.doi:10.1039/c0ee00205d
(35) Astuti,Y.;Palomares,E.;Haque,S.A.;Durrant,J.R.J.Am. Chem.Soc.2005,127,15120.doi:10.1021/ja0533444
(36) Zhang,X.F.;Shen,T.Chem.Online 1995,No.6,8.[张先付,沈 涛.化学通报,1995,No.6,8.]
(37) LópezArbeloa,I.Dyes Pigm.1983,4,211.
(38) Valdes-Aguiiera,O.;Neckers,D.C.Accounts Chem.Res. 1989,22,171.doi:10.1021/ar00161a002
(39) De,S.;Das,S.;Girigoswami,A.Spectrochim.Acta Part A 2005,61,1821.doi:10.1016/j.saa.2004.06.054
(40) Yan,Z.P.;Yu,X.X.;Zhang,Y.Y.;Jia,H.X.;Sun,Z.J.;Du,P. W.Appl.Catal.B:Environ.2014,160-161,173.
(41) Pelet,S.;Grätzel,M.;Moser,J.E.J.Phys.Chem.B 2003,107, 3215.
(42) Zhang,X.J.;Jin,Z.L.;Li,Y.X.;Li,S.B.;Lu,G.X. J.Colloid Interface Sci.2009,333,285.doi:10.1016/j. jcis.2009.01.013
(43) Zhang,X.J.;Tang,Z.Q.;Jin,Z.L.;Lü,G.X.;Li,S.B.Acta Phys.-Chim.Sin.2011,27(5),1143.[张晓杰,汤长青,靳治良,吕功煊,李树本.物理化学学报,2011,27(5),1143.]doi: 10.3866/PKU.WHXB20110511
(44) Kasche,V.;Lindqvist,L.Photochem.Photobiol.1965,4,923. doi:10.1111/php.1965.4.issue-5
(45) Shimidzu,T.;Iyoda,T.;Koide,Y.J.Am.Chem.Soc.1985, 107,35.doi:10.1021/ja00287a007
(46) Birla,L.;Cristian,A.M.;Hillebrand,M.Spectrochim.Acta Part A 2004,60,551.doi:10.1016/S1386-1425(03)00261-0
(47) Mau,A.W.H.;Johansen,O.;Sasse,W.H.F.Photochem. Photobiol.1985,41,503.doi:10.1111/php.1985.41.issue-5
(48) Heleg,V.;Willner,I.J.Chem.Soc.,Chem.Commun.1994, 2113.
(49) Gurunathan,K.;Maeuthamuthu,P.;Sastri,M.V.C.Int.J. Hydrog.Energy 1997,22,57.doi:10.1016/S0360-3199(96) 00075-4
(50) Abe,R.;Sayama,K.;Arakawa,H.Chem.Phys.Lett.2002, 362,441.doi:10.1016/S0009-2614(02)01140-5
(51) Xie,C.F.;Li,Y.X.;Peng,S.Q.;Lü,G.X.;Li,S.B.Acta Energiae Solaris Sin.2007,28(9),956.[谢称福,李越湘,彭绍琴,吕功煊,李树本.太阳能学报,2007,28(9),956.]
(52) Chatterjee,D.Catal.Commun.2010,11,336.doi:10.1016/j. catcom.2009.10.026
(53) Sreethawong,T.;Junbua,C.;Chavade,S.J.Power Sources 2009,190,513.doi:10.1016/j.jpowsour.2009.01.054
(54) Abe,R.;Sayama,K.;Arakawa,H.J.J.Photochem.Photobiol. A:Chem.2004,166,115.doi:10.1016/j.jphotochem. 2004.04.031
(55) Liu,F.S.;Ji,R.;Wu,M.;Sun,Y.M.Acta Phys.-Chim.Sin. 2007,23(12),1899.[刘福生,吉 仁,吴 敏,孙岳明.物理化学学报,2007,23(12),1899.]doi:10.3866/PKU. WHXB20071213
(56) Maia,D.L.S.;Pepe,I.;Ferreirada Silva,A.;Silva,L.A. J.Photochem.Photobiol.A:Chem.2012,243,61.doi: 10.1016/j.jphotochem.2012.06.008
(57) Zhang,G.;Choi,W.Chem.Commun.2012,48,10621.doi: 10.1039/c2cc35751h
(58) Yang,J.B.;Ganesan,P.;Teuscher,J.;Moehl,T.;Kim,Y.J.;Yi, C.Y.;Comte,P.;Pei,K.;Holcombe,T.W.;Nazeeruddin,M. K.;Hua,J.L.;Zakeeruddin,S.M.;Tian,H.;Grätzel,M. J.Am.Chem.Soc.2014,136,5722.doi:10.1021/ja500280r
(59) Cai,Z.X.;Luo,H.W.;Qi,P.L.;Wang,J.G.;Zhang,G.X.; Liu,Z.T.;Zhang,D.Q.Macromolecules 2014,47,2899.
(60) Li,H.;Wu,Y.Z.;Geng,Z.Y.;Liu,J.C.;Xu,D.D.;Zhu,W. H.J.Mater.Chem.A 2014,2,14649.
(61) Zhang,X.H.;Peng,T.Y.;Yu,L.J.;Li,R.J.;Li,Q.Q.;Li,Z. ACS Catal.2015,5,504.
(62) Zhao,W.;Hou,Y.J.;Wang,X.S.;Zhang,B.W.;Cao,Y.; Yang,R.;Wang,W.B.;Xiao,X.R.Sol.Energy Mate.Sol. Cells 1999,58,173.doi:10.1016/S0927-0248(98)00201-3
(63) Min,S.X.;Lü,G.X.Int.J.Hydrog.Energy 2012,37,10564. (64) Li,B.;Lü,G.X.J.Mol.Catal.(China)2013,27(2),181. [李 波,吕功煊.分子催化,2013,27(2),181.]
(65) Liu,X.;Li,Y.X.;Peng,S.Q.;Lu,G.X.;Li,S.B.Photochem. Photobiol.Sci.2013,12,1903.doi:10.1039/c3pp50167a
(66) Maeda,K.;Eguchi,M.;Youngblood,J.;Mallouk,T.E.Chem. Mater.2008,20,6770.doi:10.1021/cm801807b
(67) Chen,Y.J.;Mou,Z.G.;Yin,S.L.;Huang,H.;Yang,P.;Wang, X.M.;Du,Y.K.Mater.Lett.2013,107,31.doi:10.1016/j. matlet.2013.05.065
(68) Choi,S.K.;Kim,S.;Ryu,J.;Lim,S.K.;Park,H.Photochem. Photobiol.Sci.2012,11,1437.doi:10.1039/c2pp25054c
(69) Perera,V.P.S.;Senadeera,G.K.R.;Tennakone,K.S. J.Colloid Interface Sci.2003,265,428.
(70) Puangpetch,T.;Sommakettarin,P.;Chavadej,S.;Sreethawong, T.Int.J.Hydrog.Energy 2010,35,12428.doi:10.1016/j. ijhydene.2010.08.138
(71) Li,Q.Y.;Jin,Z.L.;Peng,Z.G.;Li,Y.X.;Li,S.B.;Lu,G.X. J.Phys.Chem.C 2007,111,8237.doi:10.1021/jp068703b
(72) Li,Q.Y.;Chen,L.;Lu,G.X.J.Phys.Chem.C 2007,111, 11494.doi:10.1021/jp072520n
(73) Li,Q.Y.;Lu,G.X.J.Mol.Catal.A:Chem.2007,266,75.doi: 10.1016/j.molcata.2006.10.047
(74) Liu,X.;Li,Y.X.;Peng,S.Q.;Lu,G.X.;Li,S.B.Int.J. Hydrog.Energy 2012,37,12150.doi:10.1016/j. ijhydene.2012.06.028
(75) Min,S.X.;Lu,G.X.J.Phys.Chem.C 2011,115,13938.doi: 10.1021/jp203750z
(76) Zhang,W.Y.;Li,Y.X.;Peng,S.Q.;Cai,X.Beilstein J. Nanotechnol.2014,5,801.doi:10.3762/bjnano.5.92
(77) Min,S.X.;Lu,G.X.J.Phys.Chem.C 2012,116,19644.doi: 10.1021/jp304022f
(78) Xu,J.Y.;Li,Y.X.;Peng,S.Q.;Lu,G.X.;Li,S.B.Phys. Chem.Chem.Phys.2013,15,7657.doi:10.1039/c3cp44687e
(79) Xu,J.Y.;Li,Y.X.;Peng,S.Q.Int.J.Hydrog.Energy 2015, 40,353.doi:10.1016/j.ijhydene.2014.10.150
(80) Zhang,J.;Liu,X.H.Phys.Chem.Chem.Phys.2014,16,8655. doi:10.1039/c4cp00084f
(81) Zhang,X.J.;Jin,Z.L.;Li,Y.X.;Li,S.B.;Lu,G.X.Appl. Surf.Sci.2008,254,4452.doi:10.1016/j.apsusc.2008.01.038
(82) Kataoka,Y.;Sato,K.;Miyazaki,Y.;Masuda,K.;Tanaka,H.; Naito,S.;Mori,W.Energy Environ.Sci.2009,2,397.doi: 10.1039/b814539c
(83) Wang,C.;deKrafft,K.E.;Lin,W.J.Am.Chem.Soc.2012, 134,7211.doi:10.1021/ja300539p
(84) Fateeva,A.;Chater,P.A.;Ireland,C.P.;Tahir,A.A.; Khimyak,Y.Z.;Wiper,P.V.;Darwent,J.R.;Rosseinsky,M.J. Angew.Chem.Int.Edit.2012,51,7440.doi:10.1002/ anie.201202471
(85) Kim,W.;Tachikawa,T.;Majima,T.J.Phys.Chem.C 2009, 113,10603.
(86) Jin,Z.L.;Zhang,X.J.;Li,Y.X.;Li,S.B.;Lu,G.X.Catal. Commun.2007,8,1267.doi:10.1016/j.catcom.2006.11.019
(87) Li,Y.X.;Xie,C.F.;Peng,S.Q.;Lu,G.X.;Li,S.B.J.Mol. Catal.A:Chem.2008,282,117.doi:10.1016/j. molcata.2007.12.005
(88) Guo,M.M.;Li,Y.X.;Peng,S.Q.;Lü,G.X.;Li,S.B. J.Funct.Mater.2009,5(40),802.[郭苗苗,李越湘,彭绍琴,吕功煊,李树本.功能材料,2009,5(40),802.]
(89) Li,Y.X.;Guo,M.M.;Peng,S.Q.;Lu,G.X.;Li,S.B.Int.J. Hydrog.Energy 2009,34,5629.doi:10.1016/j. ijhydene.2009.05.100
(90) Zhang,X.J.;Jin,Z.L.;Li,Y.X.;Li,S.B.;Lu,G.X.J.Power Sources 2007,166,74.doi:10.1016/j.jpowsour.2006.12.082
(91) Zeng,L.Y.;Dai,S.Y.;Wang,K.J.;Shi,C.W.;Kong,F.T.; Hu,L.H.;Pan,X.Acta Phys.Sin.2005,54(1),53. [曾隆月,戴松元,王孔嘉,史成武,孔凡太,胡林华,潘 旭.物理学报, 2005,54(1),53.]
(92) Murakoshi,K.;Kano,G.;Wada,Y.;Yanagida,S.;Miyazaki, H.;Matsumoto,M.;Murasawa,S.J.Electroanal.Chem.1995, 396,27.doi:10.1016/0022-0728(95)04185-Q
(93) Abe,R.;Hara,K.;Sayama,K.;Domen,K.;Arakawa,H. J.Photochem.Photobiol.A:Chem.2000,137,63.doi: 10.1016/S1010-6030(00)00351-8
(94) Fung,A.K.M.;Chiu,B.K.W.;Lam,M.H.W.Water Res. 2003,37,1939.doi:10.1016/S0043-1354(02)00567-5
(95) Chen,Y.S.;Li,C.;Zeng,Z.H.;Wang,W.B.;Wang,X.S.; Zhang,B.W.J.Mater.Chem.2005,15,1654.doi:10.1039/ b418906j
(96) Regazzoni,A.E.;Mandelbaum,P.;Matsuyoshi,M.;Schiller, S.;Bilmes,S.A.;Blesa,M.A.Langmuir 1998,14,868.doi: 10.1021/la970665n
(97) Ikeda,S.;Abe,C.;Torimoto,T.;Ohtani,B.J.Photochem. Photobiol.A:Chem.2003,160,61.doi:10.1016/S1010-6030 (03)00222-3
(98) Kalyanasundaram,K.;Grätzel,M.Coord.Chem.Rev.1998, 77,347.
(99) Peng,T.Y.;Ke,D.N.;Cai,P.;Dai,K.;Ma,L.;Zan,L. J.Power Sources 2008,180,498.doi:10.1016/j. jpowsour.2008.02.002
(100) Bae,E.;Choi,W.;Park,J.;Shin,H.S.;Kim,S.B.;Lee,J.S. J.Phys.Chem.B 2004,108,14093.doi:10.1021/jp047777p
(101) Jin,Z.L.;Zhang,X.J.;Lu,G.X.;Li,S.B.J.Mol.Catal.A: Chem.2006,259,275.doi:10.1016/j.molcata.2006.06.035
(102) Min,S.X.;Lu,G.X.J.Phys.Chem.C 2012,116,25415.doi: 10.1021/jp3093786
(103) Kong,C.;Min,S.X.;Lu,G.X.Int.J.Hydrog.Energy 2014, 39,4836.doi:10.1016/j.ijhydene.2014.01.089
(104) Kang,S,Z.;Chen,L.L.;Li,X.Q.;Mu,J.Appl.Surf.Sci. 2012,258,6029.doi:10.1016/j.apsusc.2012.02.118
(105) Mu.J.;Chen,L.L.;Kang,S.Z.;Li,X.Q.Chin.J.Inorg. Chem.2012,28(2),251.[穆 劲,陈丽莉,康诗钊,李向清.无机化学学报,2012,28(2),251.]
(106) Li,X.Q.;Zhang,J.;Kang,S.Z.;Li,G.D.;Mu,J.Ceram.Int. 2014,40,10171.doi:10.1016/j.ceramint.2014.02.055
(107) Zhang,W.;Xu,R.Int.J.Hydrog.Energy 2012,37,17899.doi: 10.1016/j.ijhydene.2012.08.150
(108) Zhang,X.J.;Jin,Z.L.;Li,Y.X.;Li,S.B.;Lu,G.X.J.Phys. Chem.C 2009,113,2630.doi:10.1021/jp8085717
(109) Nada,A.A.;Hamed,H.A.;Barakat,M.H.;Mohamed,N.R.; Veziroglu,T.N.Int.J.Hydrog.Energy 2008,33,3264.doi: 10.1016/j.ijhydene.2008.04.027
(110) Abe,R.;Sayama,K.;Arakawa,H.Chem.Phys.Lett.2003, 379,230.doi:10.1016/j.cplett.2003.07.026
(111) Kalyanasundaram,K.;Kiwi,J.;Grätzel,M.HeIv.Chim.Acta 1978,61,2720.
(112) Han,Z.J.;McNamara,W.R.;Eum,M.;Holland,P.L.; Eisenberg,R.Angew.Chem.Int.Edit.2012,51,1667.doi: 10.1002/anie.v51.7
(113) McCormick,T.M.;Calitree,B.D.;Orchard,A.;Kraut,N.D.; Bright,F.V.;Detty,M.R.;Eisenberg,R.J.Am.Chem.Soc. 2010,132,15480.doi:10.1021/ja1057357
(114) Lazarides,T.;McCormick,T.;Du,P.W.;Luo,G.G.;Lindley, B.;Eisenberg,R.J.Am.Chem.Soc.2009,131,9192.doi: 10.1021/ja903044n
(115) Krishnan,C.V.;Sutin,N.J.Am.Chem.Soc.1981,103,2141. doi:10.1021/ja00398a066
(116) Probst,B.;Guttentag,M.;Rodenberg,A.;Hamm,P.;Alberto, R.Inorg.Chem.2011,50,3404.doi:10.1021/ic102317u
(117) Zhu,M.S.;Li,Z.;Du,Y.K.;Mou,Z.G.;Yang,P. ChemCatChem 2012,4,112.doi:10.1002/cctc.v4.1
(118) Hori,H.;Ishihara,J.;Koike,K.;Takeuchi,K.;Ibusuki,T.; Ishitani,O.J.Photochem.Photobiol.A:Chem.1999,120, 119.doi:10.1016/S1010-6030(98)00430-4
(119) Li,B.;Lü,G.X.Acta Phys.-Chim.Sin.2013,29(8),1778. [李 波,吕功煊.物理化学学报,2013,29(8),1778.]doi: 10.3866/PKU.WHXB201305302
(120) Hong,J.D.;Wang,Y.B.;Pan,J.S.;Zhong,Z.Y.;Xu,R. Nanoscale 2011,3,4655.doi:10.1039/c1nr10628g
(121) Liu,X.;Li,Y.X.;Peng,S.Q.;Lu,G.X.;Li,S.B.Int.J. Hydrog.Energy 2013,38,11709.doi:10.1016/j. ijhydene.2013.06.095
(122) Wang,J.L.;Wang,C.;Lin,W.ACS Catal.2012,2,2630.doi: 10.1021/cs3005874
(123) Roy,N.;Sohn,Y.;Pradhan,D.ACS Nano 2013,7,2532.doi: 10.1021/nn305877v
(124) Liu,C.;Han,X.G.;Xie,S.F.;Kuang,Q.;Wang,X.;Jin,M. S.;Xie,Z.X.;Zheng,L.S.Chem.Asian J.2013,8,282.
(125) Gu,L.A.;Wang,J.Y.;Cheng,H.;Du,Y.C.;Han,X.J.Chem. Commun.2012,48,6 978.
(126) Abe,R.;Shinmei,K.;Koumura,N.;Hara,K.;Ohtani,B. J.Am.Chem.Soc.2013,135,16872.doi:10.1021/ja4048637
(127) Lee,J.;Kwak,J.;Ko,K.C.;Park,J.H.;Ko,J.H.;Park,N.; Kim,E.;Ryu,D.H.;Ahn,T.K.;Lee,J.Y.;Son,S.U.Chem. Commun.2012,48,11431.doi:10.1039/c2cc36501d
(128) Kumari,A.;Mondal,I.;Pal,U.New J.Chem.2015,39,713. doi:10.1039/C4NJ01436G
Progress in Visible-Light Photocatalytic Hydrogen Production by Dye Sensitization
LIU Xing1,2LI Yue-Xiang1,*PENG Shao-Qin1LAI Hua2
(1Department of Chemistry,Nanchang University,Nanchang 330031,P.R.China;2Department of Chemistry and Material Science,Hengyang Normal University,Hengyang 421008,Hunan Province,P.R.China)
Dye sensitization is an important strategy for broadening the excitation wavelength range of wideband-gap photocatalysts to use visible light from the sun.In this paper,the primary principle of dye-sensitized water splitting for hydrogen production was introduced,and the research progress in dye sensitizers,sensitized matrixes or supporters,the interaction between dyes and matrixes,co-catalysts for hydrogen evolution,and sacrificial electron donors were all reviewed.Moreover,the pathways of charge transmission and stability issues in dye-sensitized systems were discussed.
Photocatalytic hydrogen production;Dye sensitization;Sensitizer;Sensitized matrix; Stability
O643
10.3866/PKU.WHXB201502041www.whxb.pku.edu.cn
Received:December 15,2014;Revised:February 2,2015;Published on Web:February 4,2015.
∗Corresponding author.Email:liyx@ncu.edu.cn;Tel:+86-791-83969983.
The project was supported by the National Nature Science Foundation of China(21163012),National Key Basic Research Program of China(973) (2009CB220003),Scientific Research Foundation of Hengyang Normal University,China(14B24),and Project of Science and Technology Bureau of Hengyang City,China(2014KJ18).
国家自然科学基金(21163012),国家重点基础研究发展规划项目(973)(2009CB220003),衡阳师范学院科学基金项目(14B24)和衡阳市科学技术发展计划项目(2014KJ18)资助
