基于分子技术的土壤微生物多样性研究进展
2014-12-29谭益民何苑皞郭文平
谭益民,何苑皞,郭文平
(1. 中南林业科技大学 经济林培育与保护教育部重点实验室,湖南 长沙 410004;2.中南林业科技大学 林学院,湖南 长沙 410004;3.湖南省攸县黄丰桥国有林场,湖南 株洲 412307)
基于分子技术的土壤微生物多样性研究进展
谭益民1,2,何苑皞1,2,郭文平3
(1. 中南林业科技大学 经济林培育与保护教育部重点实验室,湖南 长沙 410004;2.中南林业科技大学 林学院,湖南 长沙 410004;3.湖南省攸县黄丰桥国有林场,湖南 株洲 412307)
土壤微生物多样性是维系土壤功能的重要因素,但是由于传统的研究方法存在一定的局限性,使得过去研究其多样性及群落结构成为一个难题。随着分子生物学方法的应用,现在可以同时对可培养微生物和不可培养微生物进行研究,解决了传统培养方法存在的问题。综述了土壤微生物多样性的分子生物学研究方法,对比了各方法的优缺点,为选择合适的方法进行土壤微生物多样性研究提供了参考。
土壤微生物多样性;分子生物学技术;研究进展
微生物作为分解者以及生产者,支撑着整个地球上的物质循环,使生命得以延续。土壤是微生物多样性最为丰富的地方,人类仅仅研究了其中的很小一部分,绝大多数还处于未知状态。土壤微生物多样性研究对于开发微生物资源、治理环境污染、维持生态服务功能以及促进土壤可持续利用等方面具有十分重要的意义。人们对土壤微生物多样性的研究最开始是利用平板计数法,但这种方法存在缺陷,如忽略了不可培养微生物,扩大了生长迅速、产孢量大的微生物数量等。因此人们又开发了许多其他的方法研究土壤微生物多样性。近年来较为热门的研究方法是以分子生物学为基础的分子技术,如变性梯度凝胶电泳、末端限制性片段长度多态性分析等。随着高通量测序技术的开发,其在土壤微生物多样性领域的应用也日趋广泛。本文就目前应用较多的几种土壤微生物多样性分子研究方法进行综述,以期为土壤微生物多样性研究提供参考。
1 土壤微生物多样性概念
土壤微生物多样性是指土壤微生物群落的种群和种间差异,它在保持土壤质量和生态系统稳定性等方面具重要意义。土壤微生物群落结构和组成的多样性与均匀性不仅提高了土壤生态系统的稳定性和和谐性,同时也提高了对抗土壤微生态环境恶化的缓冲能力[1]。对土壤微生物多样性的研究在开发超常生物资源、应对全球气候变化、治理环境污染、维持生态服务功能以及促进土壤可持续利用等方面具有重要意义。目前对土壤微生物多样性的研究主要集中在物种多样性、遗传多样性、结构多样性及功能多样性这4个方面。
2 土壤微生物多样性研究方法
随着分子生物学技术的发展,一些新的方法被应用于土壤微生物多样性研究,如:DNA重组,DNA-DNA杂交,变性梯度凝胶电泳(DGGE),温度梯度凝胶电泳(TGGE)等。
2.1 鸟嘌呤加胞嘧啶方法(G+C)
G+C方法是基于不同的微生物种类所含有的G+C丰度不相同,不同物种之间的差异在3%~5%之间这一原理[2]。G+C丰度测定的方法主要有密度梯度离心、热变性法、不同DNA样本杂交、变性DNA(单链DNA)复性动力曲线分析等。因为不同的类群可能含有相同的G+C丰度,因此这种方法的准确度不高。G+C丰度分析方法的优点在于它不会受PCR误差的影响,但它所需DNA多达 50 μg(见表 1)。
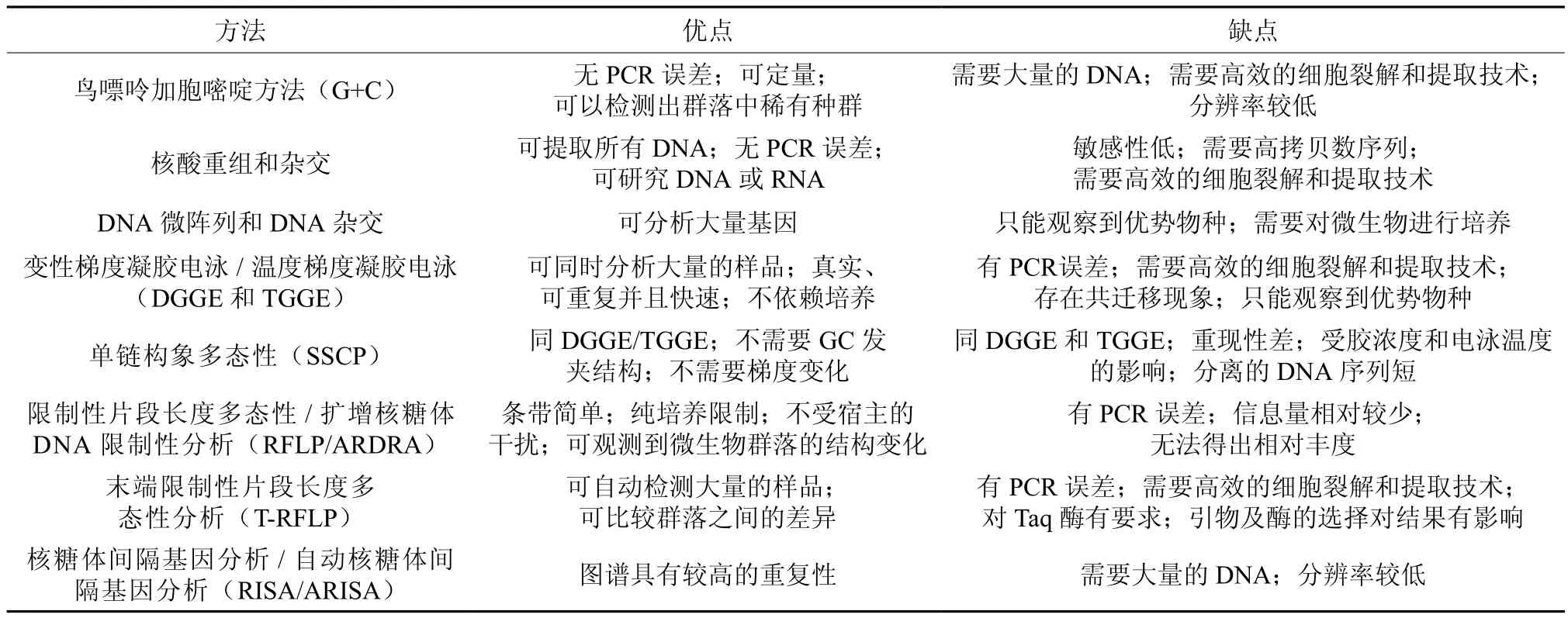
表 1 土壤微生物多样性分子研究方法的优缺点Table 1 Advantages and disadvantages of some molecular-based methods to study soil microbial diversity
Griff i ths等[3]应用该技术证明了被Cd污染的土壤中微生物群落分布与其他重金属污染(Cu,Ni,Pb,Zn)土壤中微生物群落分布存在明显差异。Nüsslein和Tiedje[4]运用G+C含量研究了夏威夷森林与牧场的不同植被下微生物多样性的变化,揭示了植物对微生物群落组成具有重要的影响。
2.2 核酸重组和杂交
核酸重组和杂交技术是基于核酸分子碱基互补配对的原理,用特异性探针与待测样品的DNA或RNA形成杂交分子的过程。这种技术通过测定DNA重组率[5]或DNA杂交动力相似度[6]来研究土壤微生物多样性。用于微生物多样性研究的探针主要有rRNA基因探针、抗性基因探针和编码代谢酶基因探针等。
核酸重组和杂交已用来研究微生物群落遗传多样性[7]以及细菌分子生态学领域[8-10]。其中荧光原位杂交(Fluorescence in Situ Hybridization, FISH)方法应用较多,并已成功用于污染环境的土壤微生物多样性研究[11]。但是也有实验表明FISH方法对于土壤样品的灵敏度较低[12]。核酸重组和杂交技术还可利用PCR扩增由mRNA反转录的cDNA研究活动微生物种群的瞬间动态。
2.3 DNA芯片
DNA芯片技术是指将数以万计的DNA探针片段有序地固定于支持物表面上,然后与标记的样品进行杂交。通过检测杂交信号来实现对生物样品的快速、平行、高效地检测或诊断。由于该法把大量DNA探针固定在狭小的空间内,因此可以实现高速度、高通量、集约化和低成本的分析。最近,DNA杂交技术和DNA芯片技术相结合用来鉴定细菌[13]和微生物多样性研究[14]。芯片既能通过所包含特殊目标基因(硝酸盐还原酶、固氮酶或萘双加氧酶基因)来提供微生物功能多样性信息,也能发现环境中的不同微生物种群。吴力游[15]在分离新的反硝化菌株、克隆亚硝酸还原酶基因的基础上构建和测试了功能基因芯片(FGA)和群落染色体芯片(CGA),建立了用这两种芯片进行的杂交检测的技术方法,并探索了它们在自然细菌群落分析上的应用。
反相基因组探针(Reverse sample genome probing, RSGP)是一种利用基因组DNA杂交来鉴定微生物的方法。RSGP的固定相是标准参照物。由于它与基因芯片的设计思路相同,只是密度比芯片小,所以RSGP也可认为是一种全基因组芯片。但使用RSGP和微阵列只能检测出最为丰富的种群,如果用基因或者DNA片段取代基因组则可以解决这一问题,并且能确定群落中的功能基因[16]。RSGP已被用于分析原油以及污染土壤中的微生物群落[17-21]。Voordouw等[20]利用RSGP技术研究了加拿大西部原油产地的采出水和腐蚀取样板中的微生物群落,发现硝酸盐还原细菌可能是金属腐蚀的原因。
2.4 基于PCR的方法
PCR通过扩增16S rDNA、18S rDNA或ITS区域对微生物进行鉴定,目前已应用于研究微生物的多样性、鉴定微生物种类以及系统发育关系[22-23]。利用PCR研究土壤微生物多样性的方法主要有变性梯度凝胶电泳(DGGE)/温度梯度凝胶电泳(TGGE),单链构象多态性(SSCP),限制性片段长度多态性(RFLP)/扩增核糖体DNA限制性分析(ARDRA)等。
2.4.1 变性梯度凝胶电泳(DGGE)/温度梯度凝胶电泳(TGGE)
变性梯度凝胶电泳(Denaturing gradient gel electrophoresis,DGGE)和温度梯度凝胶电泳(Temperature gradient gel electrophoresis,TGGE)是研究微生物多样性的两种相似方法,其中TGGE是由DGGE衍生出的技术[24]。DGGE和TGGE是利用化学变性剂或温度对DNA双链分子进行处理,使得长度相同而序列不同的DNA片段分离[24]。根据电泳条带的多寡和条带的位置可以初步辨别出样品中微生物的种类多少,粗略分析土壤样品中微生物的多样性。
DGGE和TGGE最初是用来研究DNA序列突变位点。Muyzer等[25]首次将DGGE应用到微生物遗传多样性研究上。Sachie等[26]研究了土壤中的Collimonas细菌对土壤真菌群落的影响,发现Collimonas改变了土壤真菌群落但是对微生物生物量、纤维素分解率和植物生长无影响。Korkama等[27]对挪威云杉的外生菌根研究,结果证明外生菌根的多样性及群落结构与寄主的生长率和大小有关。王奇赞等[28]采用DGGE法分析了天目山毛竹入侵阔叶林后土壤细菌群落结构的变化,结果表明入侵后土壤细菌多样性无显著变化。Cheung等[29]将TGGE与PCR相结合研究石油污染土壤中细菌多样性时,发现石油重度污染土壤中分支杆菌比轻度污染土壤中少。DGGE/TGGE还被用来研究添加不同氮肥以及复合肥对根际细菌和真菌多样性的影响。一些学者对分解代谢基因进行DGGE分析[30]获得具有某一特定功能的特殊微生物多样性的信息,如具有降解污染物功能的微生物。
2.4.2 单链构象多态性(SSCP)
单链构象多态性(Single-strand conformation polymorphism,SSCP)是另外一种对DNA扩增产物进行电泳分离分析的技术。由于单链DNA的构象差异使得它们在聚丙烯酰胺凝胶上的迁移率发生变化,因此可将不同的单链DNA分离开[31-32]。SSCP与DGGE具有相同的局限性,并且SSCP技术易受胶浓度和电泳温度等条件的影响。
与DGGE/TGGE相似,SSCP最初是用来研究人类DNA的基因多态性[33]。Lee等[32]首次将SSCP技术应用到环境样品的微生物群落多样性分析中。Stach[34]利用PCR-SSCP技术研究土壤细菌多样性。SSCP还被用于研究细菌群落的演替[35],根际微生物群落[36-37],厌氧发酵罐中细菌群落的变化[38]以及根际丛枝菌根真菌[39-40]。
2.4.3 限制性片段长度多态性(RFLP)/扩增核糖体DNA限制性分析(ARDRA)
限制性片段长度多态性(Restriction fragment length polymorphism,RFLP)方法和扩增核糖体DNA限制性分析(Amplif i ed ribosomal DNA restriction analysis,ARDRA)方法也常用于微生物群落的分析,该方法依赖于DNA多态性。RFLP广泛的应用于新物种的发现、微生物分类及鉴定、微生物遗传多样性的检测和细菌群落结构研究[41]等。RFLP是一种十分有效的观察微生物群落结构变化的方法[42]。张于光等利用RFLP方法对三江源地区不同的土壤中固氮微生物进行了研究,发现不同植物类型的土壤中分别具有1-2个优势种[43]。Sandaa等[44]用RFLP方法研究了重金属污染对土壤中可培养细菌和总细菌群落结构的影响,结果发现增加重金属浓度会降低细菌群落的多样性。Moffett等[45]利用ARDRA技术发现农田中Zn污染土壤中的细菌群落多样性低于非污染土壤。
2.4.4 末端限制性片段长度多态性分析(T-RFLP)
末端限制性片段长度多态性分析(Terminaterestriction length fragment polymorphism,T-RFLP)解决了RFLP的一些缺陷。它的基本原理与RFLP相同,不同之处是它使用了荧光染料标记的PCR引物(TET或6-FAM),在计算机程序自动分析时仅分析荧光标记的末端限制片段[46]。T-RFLP简化了条带,由于它的每一个条带代表了一种分类单位或者核糖体基因型因此它提供了多样性的信息,可以用来分析复杂群落[47]。
T-RFLP用来研究细菌群落在空间和时间上的变化[48]、细菌群落多样性[49-50]。Tonin等[51]利用T-RFLP技术观察和监测种群以及评估在重金属污染土壤中芦苇堇菜(Viola calaminaria)根际的丛枝菌根真菌多样性。张慧[52]利用T-RFLP研究了不同利用方式对红壤坡地微生物多样性的影响,发现不同利用方式对红壤坡地中微生物多样性差异不显著。有些学者认为利用T-RFLP研究微生物多样性无法得到准确结果,如Dunbar等[53]所用的统计数据与T-RFLP所产生的DNA条带相矛盾,并且四种不同类型土样之间的多样性并未发现明显的差异。
2.4.5 核糖体基因间隔区分析(RISA)/自动核糖体间隔区基因分析(ARISA)
核糖体基因间隔区分析(Ribosomal inter genic spacer analysis, RISA)和自动核糖体间隔区基因分析(Automated ribosomal intergenic spacer analysis,ARISA),都是DNA指纹技术。RISA和ARISA方法是用PCR扩增16S和23S核糖体亚基中的间隔区(ISR),然后电泳分离。由于ISR长度和序列的异质性使得ISR可以区分不同的细菌菌株以及亲缘关系较近的种群[54]。RISA和ARISA的区别在于,RISA运用银染显色技术检测序列多态性,而ARISA中上游引物用荧光标记并进行自动检测。这两种方法都能提供高重复性的细菌群落谱但是RISA需要大量的DNA并耗时,而银染显色技术敏感度不高,导致结果偏低。ARISA提高了该方法的灵敏度,降低了时耗,但是它仍然受PCR的限制。
目前RISA被用于土壤、植物根际、污染土壤以及接种后微生物多样性对比。Ranjard等[55]用RISA研究了Hg对细菌群落结构的影响,发现在Hg污染胁迫下,耐Hg细菌增多并且整个细菌群落多样性降低。陈颖等[56]利用荧光定量PCR和ARISA对非根际土壤和根际土壤中细菌和真菌的数量及群落结构进行了分析,研究结果表明不同植物物种可以通过根系影响土壤微生物群落组成。Hanene等[57]使用DGGE和ARISA方法对经过堆肥处理的农田土壤细菌群落和多样性进行研究,发现不同堆肥处理的农田土壤中的细菌群落差异不明显,同时他们得出结论DGGE进行细菌鉴定较准确,而ARISA更适用于优势群落分析。
2.5 高通量测序技术
高 通 量 测 序 技 术(High-throughput sequencing)又称“下一代”测序技术(Nextgeneration sequencing technology)[58]或深度测序(Deep sequencing)[59]。高通量测序技术以能一次对几十万到几百万条DNA分子进行序列测定为标志,这使得对一个物种的转录组和基因组进行细致全面的分析成为可能。
高通量测序技术是相对于传统的Sanger测序技术而言的。它与传统的Sanger测序技术比较其优点主要有一下几个方面:①高通量测序利用芯片进行大规模平行测序,极大的节省了时间;②高通量测序具有定量功能,可反映样品中DNA的丰度;③成本低廉,利用高通量测序技术进行人类基因组测序,耗资不到传统Sanger测序法的1%[60];④实现了边合成边测序,不需要等到测序完成后再上机。
2.5.1 高通量测序技术种类
高通量测序平台的代表是罗氏公司(Roche)的 454测 序 仪(Roch GS FLX sequencer),Illumina公司的Solexa基因组分析仪(Illumina Genome Analyzer)和ABI的SOLiD测序仪(ABI SOLiD sequencer)。高通量测序常用方法的优缺点见表2。
(1)Genome Sequencer FLX (GS FLX) 测序系统
Genome Sequencer FLX (GS FLX)测序系统是罗氏诊断公司(Roche)开发的第二代测序平台[61]。GS FLX测序是一种基于焦磷酸测序,并依靠生物发光进行DNA序列分析的新技术。GS FLX的原理简单来说就是利用DNA聚合酶、ATP硫酸化酶、荧光素酶和双磷酸酶的协同作用,将PCR反应每一个碱基(dNTP)的延伸与一次荧光信号的释放偶联起来,通过记录荧光信号的有无和强度,达到实时测定DNA序列的目的。GS FLX测序的流程大体是样品处理、文库制备、emPCR、反应板准备、上机测序。其核心是“一个片段=一个磁珠=一条读长(one fragment=one bead=one read)”。也就是一个300~800 bp的DNA片段通过接头(adaptor)与磁珠相连,即每个磁珠都只结合一条DNA片段,每个磁珠只产生一条读长,并通过GS FLX系统进行分析。目前最新的GS FLX系统,一次运行可获得一百多万个读长片段,高质量读长达到400 bp,读取超过5亿个碱基信息。GS FLX测序系统的特点主要是可检测的物质范围较大、研究定量化以及实现了真正意义的高通量

表2 高通量测序各方法优缺点Table 2 Advantages and disadvantages of different methods of high-throughput sequencing
(2)Illumina Genome Analyzer测序系统
Illumina公司的新一代测序仪Genome Analyzer最早由Solexa公司研发[62]。与GS FLX测序系统一样,Illumina Genome Analyzer测序系统通过可逆性终止的SBS(边合成边测序)技术对待测的模板DNA进行测序。不同是Illumina Genome Analyzer测序系统直接在dNTP上连接荧光基团和阻断基团,通过“去阻断—延伸—激发荧光—切割荧光基团—去阻断”这样一个循环的方法来依次读取目的DNA上的碱基排列顺序,而不是采用焦磷酸氧化荧光素的形式激发光信号。Illumina Genome Analyzer测序的大体流程是样品处理、文库制备、DNA片段在Cluster Station上成簇扩增、DNA簇在Genome Analyzer上边合成边测序、Paired-End Module、数据分析。
(3)ABI SOLiD sequencer测序系统
SOLiD全 称 为supported oligo ligation detetion,它的独特之处在于以四色荧光标记寡核苷酸的连续连接反应为基础,取代了传统的聚合酶连接反应,可对单拷贝DNA片段进行大规模扩增和高通量并行测序[63]。SOLiD测序既没有DNA聚合酶合成过程中的错配问题,又有SOLiD特有的“双碱基编码原理”提供的纠错机制,这使得每个碱基可被判读2次,系统准确性达99.99%,因此SOLiD的系统准确性大大领先于其他测序平台。由于SOLiD乳化PCR过程中的微珠仅1 μm比GS FLX系统要小得多,因此每张玻片能容纳更多的微珠,在同一系统中可具有更高的通量,单次运行能得到的最大数据量为300 Gb。ABI SOLiD sequencer测序的流程是文库制备、乳液PCR/ 微珠富集、微珠沉积、连接测序、数据分析。
2.5.2 高通量测序系统的应用
高通量测序主要应用于De novo测序、基因深度测序、转录组深度测序、数字表达谱、染色质免疫沉淀、甲基化分析等[64]。高通量测序在各领域的微生物多样性研究上也有应用[65-70]。其最开始是被应用于矿井中环境微生物多样性的研究[71]。He[72]等人利用高通量测序技术对土壤微生物群落结构的研究表明随着CO2升高群落结构产生显著变化。Nacke[73]等人利用454测序技术对比了不同管理模式下德国森林和草地土壤细菌的群落结构,发现不同管理模式下土壤的细菌群落结构具有显著差异,并且细菌群落结构受树种和土壤pH的影响较大。Lihua Lu等[74]人对土豆连作环境下土壤真菌进行454测序,结果表明Sordariales和Hypocreales真菌受连作的影响较大。Chunhong Mao等[75]利用GS FLX技术研究土壤中苜蓿中华根瘤菌(Sinorhizobium meliloti)时,发现了20多种新的基因,并对这些基因的功能进行了分析。Hollister[76]等研究高盐生态系统中微生物多样性时发现,在高盐环境中微生物群落的多样性仍然十分丰富,并且这些微生物与样点的含水量、磷含量、有机碳总含量以及pH具有密切的关系。McLellan[77]等研究污水处理厂的微生物多样性,结果表明污水微生物群落代表了一种独特的群落结构,这种群落结构由人类排泄微生物以及环境中特定微生物的丰富度所决定。Mulan Dai等[78]人利用GS FLX技术研究不同土壤中丛枝菌根(AM)真菌的多样性,发现AM真菌具有自我恢复能力并且农田土壤中的AM真菌具有较高的丰富度。Stéphane等[79]利用 Illumina Genome Analyzer研究不同土层的微生物群落发现有机质层与矿质层的微生物有明显差别,有机质层以细菌、脊索动物类、节肢动物以及子囊菌为主,而矿质层则以古生菌为主。
近年来国际上使用高通量测序研究土壤微生物多样性及群落结构的研究者越来越多,发表的文章近300篇,其中大部分使用的是GS FLX系统。可见GS FLX系统以读长大、时间短的优势成为目前探索土壤微生物多样性的主流手段。而Illumina系统以其数据量大、成本低的特点也将成为今后土壤微生物多样性研究的主要手段之一。
2.6 第三代测序技术
第三代测序技术是以单分子实时测序(single-molecule sequencing)和纳米孔为标志的测序技术。第三代测序平台主要有Helicos的Heliscope单分子测序仪、Pacif i c Bioscience的SMRT技术、Oxford Nanopore Technologies公司的纳米孔单分子测序技术和Life Technologies公司的FRET测序技术。第三代测序技术可解决第二代测序技术中PCR产生误差以及读长偏短的缺陷,同时它还可以直接测RNA序列以及甲基化的DNA序列。但是研究人员仍对该技术保持谨慎态度,主要是由于其错误率相对较高。目前第三代测序技术还主要用于医学及相关领域的研究[80-82]。相信在不久的将来,第三代测序技术将会更加的成熟和完善并在土壤微生物领域得到应用。
3 展 望
土壤是微生物的大本营,要全面了解土壤微生物的群落结构以及他们的功能,尚需研究人员进一步探索不同环境下土壤微生物的多样性,并交叉利用地质学、地球化学、分析化学、微生物学、分子生物学等多学科研究方法,只有综合分析各项研究结果才能获得土壤微生物群落结构和功能的全貌。分子技术克服了纯培养的缺陷,丰富了我们对土壤微生物多样性的认识。本文介绍了几种常见的基于分子技术的土壤微生物多样性研究方法。尽管这些技术可以获得不可培养微生物的信息,但是每一种方法都仅能提供土壤微生物多样性的一部分信息且他们自身都存在的缺陷,具有有一定的局限性。因此,发展新的技术研究土壤微生物多样性极具挑战性。目前我们并不知道一克土壤里到底存在多少微生物,也无法确定哪一种研究多样性的方法更好。所以在研究土壤微生物多样性时最好同时运用几种方法,以获得更多的信息。另外,目前对植物-微生物-土壤之间的相互作用研究较多,但是生物-化学-物理因素之间的相互作用我们却知之甚少。如果一个新的技术能应用在该领域,我们则对微生物及其环境之间的关系会有一个更清晰的视野。
现代分子生物学技术的发展使我们看到了揭开土壤微生物世界秘密的希望,同时也给我们提供了更广阔的研究空间,现代分子生物技术与其他方法有机结合一定会大力推进土壤微生物多样性研究。
[1] 焦晓丹,吴凤芝. 土壤微生物多样性研究方法的进展[J]. 土壤通报, 2004, (6): 789-792.
[2] Tiedje J M, Asuming-Brempong S, Nüsslein K,et al. Opening the black box of soil microbial diversity[J]. Applied Soil Ecology.1999, 13(2): 109-122.
[3] Griff i ths B S, Dı́az-Raviña M, Ritz K,et al.Community DNA hybridisation and %G+C prof i les of microbial communities from heavy metal polluted soils[J]. FEMS Microbiology Ecology.1997, 24(2): 103-112.
[4] Nüsslein K, Tiedje J M. Soil Bacterial Community Shift Correlated with Change from Forest to Pasture Vegetation in a Tropical Soil[J]. Applied and Environmental Microbiology. 1999,65(8): 3622-3626.
[5] Theron J, Cloete T E. Molecular Techniques for Determining Microbial Diversity and Community Structure in Natural Environments[J]. Critical Reviews in Microbiology. 2000, 26(1):37-57.
[6] Griffiths B S, Ritz K, Ebblewhite N,et al. Soil microbial community structure: Effects of substrate loading rates[J]. Soil Biology and Biochemistry. 1998, 31(1): 145-153.
[7] Torsvik V, Sørheim R, Goksøyr J. Total bacterial diversity in soil and sediment communities—A review[J]. Journal of Industrial Microbiology and Biotechnology. 1996, 17(3): 170-178.
[8] Schramm A, Larsen L H, Revsbech N P,et al. Structure and function of a nitrifying biof i lm as determined by microelectrodes and fluorescent oligonucleotide probes[J]. Water Science and Technology. 1997, 36(1): 263-270.
[9] Guo C, Sun W, Harsh J B,et al. Hybridization Analysis of Microbial DNA from Fuel Oil-Contaminated and Noncontaminated Soil[J]. Microbial Ecology. 1997, 34(3): 178-187.
[10] Clegg C D, Ritz K, Griffiths B S. %G+C profiling and cross hybridisation of microbial DNA reveals great variation in belowground community structure in UK upland grasslands[J]. Applied Soil Ecology. 2000, 14(2): 125-134.
[11] Yang Y, Zeyer J. Specific Detection of Dehalococcoides Species by Fluorescence In Situ Hybridization with 16S rRNATargeted Oligonucleotide Probes[J]. Applied and Environmental Microbiology. 2003, 69(5): 2879-2883.
[12] Hahn D, Amann R I, Ludwig W,et al.Detection of microorganisms in soil after in situ hybridization with rRNA-targeted,fluorescently labelled oligonucleotides.[J]. Journal of general microbiology. 1992, 138(5): 879-887.
[13] Cho J, Tiedje J M. Bacterial Species Determination from DNADNA Hybridization by Using Genome Fragments and DNA Microarrays[J]. Applied and Environmental Microbiology. 2001,67(8): 3677-3682.
[14] Greene E A, Voordouw G. Analysis of environmental microbial communities by reverse sample genome probing[J]. Journal of Microbiological Methods. 2003, 53(2): 211-219.
[15] 吴力游. 建立用于环境及土壤微生物群落分析的基因芯片技术[D]. 湖南农业大学, 2001.
[16] Greene E A, Voordouw G. Analysis of environmental microbial communities by reverse sample genome probing[J]. Journal of Microbiological Methods. 2003, 53(2): 211-219.
[17] Voordouw G, Voordouw J K, Karkhoff-Schweizer R R,et al. Reverse Sample Genome Probing, a New Technique for Identification of Bacteria in Environmental Samples by DNA Hybridization, and Its Application to the Identification of Sulfate-Reducing Bacteria in Oil Field Samples[J]. Applied and Environmental Microbiology. 1991, 57(11): 3070-3078.
[18] Voordouw G, Shen Y, Harrington C S,et al. Quantitative Reverse Sample Genome Probing of Microbial Communities and Its Application to Oil Field Production Waters[J]. Applied and Environmental Microbiology. 1993, 59(12): 4101-4114.
[19] Shen Y, Stehmeier L G, Voordouw G. Identification of Hydrocarbon-Degrading Bacteria in Soil by Reverse Sample Genome Probing[J]. Applied and Environmental Microbiology.1998, 64(2): 637-645.
[20] Hubert C, Shen Y, Voordouw G. Composition of Toluene-Degrading Microbial Communities from Soil at Different Concentrations of Toluene[J]. Applied and Environmental Microbiology. 1999, 65(7): 3064-3070.
[21] Greene E A, Kay J G, Jaber K,et al.Composition of Soil Microbial Communities Enriched on a Mixture of Aromatic Hydrocarbons[J]. Applied and Environmental Microbiology.2000, 66(12): 5282-5289.
[22] Pace N R. New perspective on the natural microbial world:molecular microbial ecology[J]. ASM News. 1996, 62(9): 463-470.
[23] Pace N R. A molecular view of microbial diversity and the biosphere[J]. Science. 1997, 276(5313): 734-740.
[24] Muyzer G. DGGE/TGGE a method for identifying genes from natural ecosystems[J]. Current Opinion in Microbiology. 1999,2(3): 317-322.
[25] Muyzer G, De Waal E C, Uitterlinden A G. Prof i ling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplif i ed genes coding for 16S rRNA.[J]. Applied and Environmental Microbiology. 1993,59(3).
[26] Höppener-Ogawa S, Leveau J H J, Hundscheid M P J,et al.Impact of Collimonas bacteria on community composition of soil fungi[J]. Environmental Microbiology. 2009, 11(6): 1444-1452.
[27] Korkama T, Pakkanen A, Pennanen T. Ectomycorrhizal community structure varies among Norway spruce (Picea abies)clones[J]. New Phytologist. 2006, 171(4): 815-824.
[28] 王奇赞,徐秋芳,姜培坤,等. 天目山毛竹入侵阔叶林后土壤细菌群落16S rDNA V3区片段PCR的DGGE分析[J]. 土壤学报. 2009, (4): 662-669.
[29] Cheung P, Kinkle B K. Mycobacterium Diversity and Pyrene Mineralization in Petroleum-Contaminated Soils[J]. Applied and Environmental Microbiology. 2001, 67(5): 2222-2229.
[30] Knief C, Lipski A, Dunfield P F. Diversity and Activity of Methanotrophic Bacteria in Different Upland Soils[J]. Applied and Environmental Microbiology. 2003, 69(11): 6703-6714.
[31] Schwieger F, Tebbe C C. A New Approach To Utilize PCRSingle-Strand-Conformation Polymorphism for 16S rRNA Gene-Based Microbial Community Analysis[J]. Applied and Environmental Microbiology. 1998, 64(12): 4870-4876.
[32] Lee D H, Zo Y G, Kim S J. Nonradioactive method to study genetic profiles of natural bacterial communities by PCR-single-strand-conformation polymorphism.[J]. Applied and Environmental Microbiology. 1996, 62(9): 3112-3120.
[33] Orita M, Suzuki Y, Sekiya T,et al. Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction[J]. Genomics. 1989, 5(4): 874-879.
[34] Stach J E M, Bathe S, Clapp J P,et al. PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification methods[J]. FEMS Microbiology Ecology. 2001, 36(2-3): 139-151.
[35] Peters S, Koschinsky S, Schwieger F,et al.Succession of Microbial Communities during Hot Composting as Detected by PCR-Single-Strand-Conformation Polymorphism-Based Genetic Prof i les of Small-Subunit rRNA Genes[J]. Applied and Environmental Microbiology. 2000, 66(3): 930-936.
[36] Schwieger F, Tebbe C C. A New Approach To Utilize PCRSingle-Strand-Conformation Polymorphism for 16S rRNA Gene-Based Microbial Community Analysis[J]. Applied and Environmental Microbiology. 1998, 64(12): 4870-4876.
[37] Schmalenberger A, Schwieger F, Tebbe C C. Effect of Primers Hybridizing to Different Evolutionarily Conserved Regions of the Small-Subunit rRNA Gene in PCR-Based Microbial Community Analyses and Genetic Profiling[J]. Applied and Environmental Microbiology. 2001, 67(8): 3557-3563.
[38] Zumstein E, Moletta R, Godon J. Examination of two years of community dynamics in an anaerobic bioreactor using fluorescence polymerase chain reaction (PCR) single-strand conformation polymorphism analysis[J]. Environmental Microbiology. 2000, 2(1): 69-78.
[39] Simon L, Lévesque R C, Lalonde M. Identification of endomycorrhizal fungi colonizing roots by fluorescent singlestrand conformation polymorphism-polymerase chain reaction.[J]. Applied and Environmental Microbiology. 1993, 59(12):4211-4215.
[40 Kjøller R, Rosendahl S. Detection of arbuscular mycorrhizal fungi (Glomales) in roots by nested PCR and SSCP (Single Stranded Conformation Polymorphism)[J]. Plant and Soil. 2000,226(2): 189-196.
[41] Greene E A, Voordouw G. Analysis of environmental microbial communities by reverse sample genome probing[J]. Journal of Microbiological Methods. 2003, 53(2): 211-219.
[42] Acinas S G, Rodrı́guez-Valera F, Pedrós-Alió C. Spatial and temporal variation in marine bacterioplankton diversity as shown by RFLP fi ngerprinting of PCR amplif i ed 16S rDNA[J]. FEMS Microbiology Ecology. 1997, 24(1): 27-40.
[43] 张于光,王慧敏,李迪强,等. 三江源地区不同植被土壤固氮微生物的群落结构研究[J]. 微生物学报. 2005, (3): 420-425.
[44] Sandaa R A, Torsvik V, Enger Ø. Inf l uence of long-term heavymetal contamination on microbial communities in soil[J]. Soil Biology and Biochemistry. 2001, 33(3): 287-295.
[45] Moffett B F, Nicholson F A, Uwakwe N C,et al.Zinc contamination decreases the bacterial diversity of agricultural soil[J]. FEMS Microbiology Ecology. 2003, 43(1): 13-19.
[46] Pandey J, Sood S S, Jain R K. Terminal restriction fragment length polymorphism (T-RFLP) analysis: Characterizing the unseen[J]. Indian Journal of Microbiology. 2007, 47(1): 90-91.
[47] Liu W T, Marsh T L, Cheng H,et al.Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA.[J]. Applied and Environmental Microbiology. 1997, 63(11): 4516-4522.
[48] Lukow T, Dunf i eld P F, Liesack W. Use of the T-RFLP technique to assess spatial and temporal changes in the bacterial community structure within an agricultural soil planted with transgenic and non-transgenic potato plants[J]. FEMS Microbiology Ecology.2000, 32(3): 241-247.
[49] Clement B G, Kehl L E, Debord K L,et al. Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities[J]. Journal of Microbiological Methods. 1998, 31(3): 135-142.
[50] Moeseneder M M, Arrieta J M, Muyzer G,et al.Optimization of Terminal-Restriction Fragment Length Polymorphism Analysis for Complex Marine Bacterioplankton Communities and Comparison with Denaturing Gradient Gel Electrophoresis[J].Applied and Environmental Microbiology. 1999, 65(8): 3518-3525.
[51] Tonin C, Vandenkoornhuyse P, Joner E J,et al.Assessment of arbuscular mycorrhizal fungi diversity in the rhizosphere of Viola calaminaria and effect of these fungi on heavy metal uptake by clover[J]. Mycorrhiza. 2001, 10(4): 161-168.
[52] 张 慧,袁红朝,朱亦君,等. 不同利用方式对红壤坡地微生物多样性和硝化势的影响[J]. 生态学杂志,2011,(6):1169-1176.
[53 Dunbar J, Ticknor L O, Kuske C R. Assessment of Microbial Diversity in Four Southwestern United States Soils by 16S rRNA Gene Terminal Restriction Fragment Analysis[J]. Applied and Environmental Microbiology. 2000, 66(7): 2943-2950.
[54] Fisher M M, Triplett E W. Automated Approach for Ribosomal Intergenic Spacer Analysis of Microbial Diversity and Its Application to Freshwater Bacterial Communities[J]. Applied and Environmental Microbiology. 1999, 65(10): 4630-4636.
[55] Ranjard L, Nazaret S, Gourbière F,et al.A soil microscale study to reveal the heterogeneity of Hg(II) impact on indigenous bacteria by quantif i cation of adapted phenotypes and analysis of community DNA fi ngerprints[J]. FEMS Microbiology Ecology.2000, 31(2): 107-115.
[56] 陈 颖,李肖肖,应娇妍,等. 内蒙草原不同植物功能群及物种对土壤微生物组成的影响[J]. 生物多样性, 2012,(1):59-65.
[57] Cherif H, Ouzari H, Marzorati M,et al. Bacterial community diversity assessment in municipal solid waste compost amended soil using DGGE and ARISA fi ngerprinting methods[J]. World Journal of Microbiology and Biotechnology. 2008, 24(7): 1159-1167.
[58] Schuster S C. Next-generation sequencing transforms today’s biology[J]. 2008, 5(1): 16-18.
[59] Sultan M, Schulz M H, Richard H,et al.A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome[J]. Science. 2008, 321(5891): 956-960.
[60] Wheeler D A, Srinivasan M, Egholm M,et al.The complete genome of an individual by massively parallel DNA sequencing[J]. 2008, 452(7189): 872-876.
[61] Margulies M, Egholm M, Altman W E,et al.Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature.2005, 437(7057): 376-380.
[62] Bentley D R, Balasubramanian S, Swerdlow H P,et al. Accurate whole human genome sequencing using reversible terminator chemistry[J]. Nature. 2008, 456(7218): 53-59.
[63] Smith D R, Quinlan A R, Peckham H E,et al.Rapid wholegenome mutational profiling using next-generation sequencing technologies[J]. Genome Research. 2008, 18(10): 1638-1642.
[64] Diguistini S, Liao N, Platt D,et al.De novo genome sequence assembly of a fi lamentous fungus using Sanger, 454 and Illumina sequence data.[J]. Genome Biol. 2009, 10(9): R94.
[65] Turnbaugh P J, Ley R E, Mahowald M A,et al.An obesity-associated gut microbiome with increased capacity for energy harvest[J]. Nature. 2006, 444(7122): 1027-1031.
[66] Dethlefsen L, Huse S, Sogin M L,et al.The Pervasive Effects of an Antibiotic on the Human Gut Microbiota, as Revealed by Deep 16S rRNA Sequencing[J]. PLoS Biol. 2008, 6(11): e280.
[67] Costello E K, Lauber C L, Hamady M,et al.Bacterial Community Variation in Human Body Habitats Across Space and Time[J]. Science. 2009, 326(5960): 1694-1697.
[68] Qin J, Li R, Raes J,et al.A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature. 2010,464(7285): 59-65.
[69] Zaura E, Keijser B, Huse S,et al.Def i ning the healthy “core microbiome” of oral microbial communities[J]. BMC Microbiology. 2009, 9(1): 1-12.
[70] Edwards R, Rodriguez-Brito B, Wegley L,et al.Using pyrosequencing to shed light on deep mine microbial ecology[J].BMC Genomics. 2006, 7(1): 1-13.
[71] Edwards R, Rodriguez-Brito B, Wegley L,et al.Using pyrosequencing to shed light on deep mine microbial ecology[J].BMC Genomics. 2006, 7(1): 1-13.
[72] He Z, Xu M, Deng Y,et al. Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2: Changes in the soil microbial community at elevated CO2[J]. Ecology Letters. 2010, 13(5):564-575.
[73] Thürmer A, Wollherr A, Will C,et al.Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils.[J]. PLoS One. 2011, 6(2): e17000.
[74] Lu L, Yin S, Liu X,et al. Fungal networks in yieldinvigorating and -debilitating soils induced by prolonged potato monoculture[J]. Soil Biology and Biochemistry. 2013, 65(0):186-194.
[75] Mao C, Evans C, Jensen R,et al. Identification of new genes in Sinorhizobium meliloti using the Genome Sequencer FLX system[J]. BMC Microbiology. 2008, 8(1): 1-8.
[76] Hollister E B, Engledow A S, Hammett A J M,et al.Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments[J]. 2010, 4(6): 829-838.
[77] Mclellan S L, Huse S M, Mueller-Spitz S R,et al.Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent[J]. Environmental Microbiology. 2010, 12(2): 378-392.
[78] Dai M, Bainard L D, Hamel C,et al.Impact of land use on arbuscular mycorrhizal fungal communities in rural Canada[J].Applied and Environmental Microbiology. 2013.
[79] Uroz S, Ioannidis P, Lengelle J,et al.Functional Assays and Metagenomic Analyses Reveals Differences between the Microbial Communities Inhabiting the Soil Horizons of a Norway Spruce Plantation[J]. PLoS ONE. 2013, 8(2): e55929.
[80] Dancey J, Bedard P, Onetto N,et al.The Genetic Basis for Cancer Treatment Decisions[Z]. Cell Press, 2012: 148, 409-420.
[81] Loomis E W, Eid J S, Peluso P,et al.Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene[J]. Genome Research. 2012.
[82] Smith C, Wang Q, Chin C,et al.Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia.[J]. Nature. 2012, 485(7397): 260-263.
Research progress on soil microbial diversity based on molecular techniques
TAN Yi-min1,2, HE Yuan-hao1,2, GUO Wen-ping3
(1.Key Lab. for Non-wood Forest Cultivation and Conservation Supported by China Ministry of Education, Central South University of Forestry and Technology, Changsha 410004, Hunan, China; 2.College of Forestry, Central South University of Forestry and Technology,Changsha 410004, Hunan, China; 3. Hunan Huangfengqiao State-owned Forest Farm, Zhuzhou 412307, Hunan, China)
Microbial diversity within the soil is crucial to many functions but the traditional research methods had some limitations thus making the studies of the diversity and community structure to be diff i cult problems. With the application of molecular biological methods, it is now possible to detect both culture-able and un-culture-able microbial species and solve the problems of traditional methods. This review was focused on the molecular methods of soil microbial diversity and the advantages and disadvantages of these methods were compared, thus providing reference for appropriate method for microbial diversity research.
soil microbial diversity;molecular biotechnology;research progress
S718
A
1673-923X(2014)10-0001-09
2014-06-17
基金项目:林业公益性行业科研专项项目(201004014);中南林业科技大学研究生科技创新基金资助项目(2010bx02)
谭益民(1962-),男,湖南湘潭人,教授,博士,博导,主要从事林业及旅游相关研究
何苑皞(1983-),女,湖南株洲人,博士研究生,主要从事森林微生物方面的研究;E-mail:heyuanhao218@gmail.com
[本文编校:吴 彬]
