臭氧在湿法冶金领域的研究进展
2014-12-26田庆华王恒利辛云涛郭学益
田庆华,王恒利,辛云涛,郭学益
(中南大学冶金与环境学院,长沙 410083)
臭氧(O3)是氧气的同素异形体,由3个氧原子组成,在20°C时半衰期为40 min,是一种不稳定的淡紫色气体.臭氧具有极强的氧化能力,在水中的氧化还原电位(2.07 V)仅次于氟而居第二位[1-2].臭氧自1785年发现以来,作为一种强氧化剂、消毒剂、催化剂等已广泛应用于环保、食品保鲜、制药、精细化工等各个方面,并主要集中在有机物的氧化处理领域.
近年来,在臭氧氧化处理含氰废水的基础上,研究者继续探寻臭氧在其他湿法冶金领域的应用,提出了将臭氧应用于硫化物浸出和金属氧化物制备的工艺.臭氧氧化法具有氧化效率高、选择性好、反应速度快等优点,且产物氧气不会引起二次污染,反应结束后可自行分解而没有残留,符合当今清洁生产的发展要求和趋势.相比湿法冶金过程中的传统氧化剂,臭氧氧化既可避免高锰酸钾、氯酸钠、二氧化锰等氧化剂的加入引入新的杂质,也避免了双氧水等氧化剂的加入使溶液体系过度膨胀的问题,同时相比空气氧化,臭氧氧化效率更高,在常温常压下即可进行,是一种新颖、高效的清洁冶金工艺.
1 基本原理
作为一种强氧化剂,臭氧可以将一些低价态的物质氧化为高价态,在此过程中发生的反应为氧化还原反应,其特征就是在反应中有电子从一种物质转移到另外一种物质上.但是,由于反应体系酸碱性不同,臭氧在水溶液中发生的氧化还原反应也不相同,分别为[3]:
在酸性溶液中:
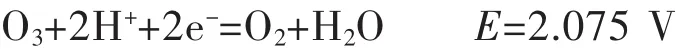
在碱性溶液中:
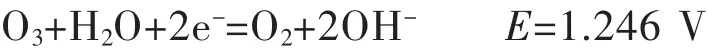
由上述反应可知,臭氧在酸性和碱性溶液中的氧化还原电位都很高,大多数的硫化物和低价金属离子都很容易被臭氧氧化.
目前,臭氧在湿法冶金领域的研究主要集中在难冶金属硫化物浸出、金属氧化物制备和含氰废水处理等方面.这些金属硫化物主要为难处理含金资源、方铅矿、黄铜矿、黝铜矿和辉铜矿等;而利用臭氧制备的金属氧化物主要为钴氧化物、羟基氧化镍、银氧化物和二氧化锰等.
1.1 难冶金属硫化物浸出
采用臭氧氧化浸出硫化矿物时,臭氧将硫化物中的硫氧化,破坏硫化矿物的结构,从而使金属得以释放进入浸出液.根据氧化程度及矿物的不同,浸出过程可能会产生2种结果:①金属硫化物中的硫元素被氧化为单质硫;②单质硫继续氧化生成硫酸根,其反应如下:
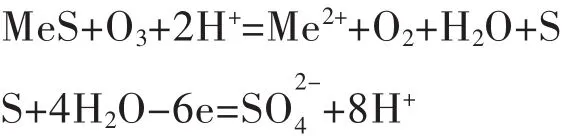
采用臭氧氧化浸出难冶金属硫化矿物的工艺,避免了高温高压操作,对设备要求不高,成本低,投资省,效益高.控制工艺条件使浸出产物为单质硫,可以避免火法系统SO2的产生,具有对环境污染小,回收价值高的优点.
1.2 金属氧化物制备
在金属氧化物制备过程中,常常采用化学沉淀法制备氧化物前驱体,再将前驱体沉淀加热分解得到相应的金属氧化物.近年来,大量研究采用臭氧作为“沉淀剂”氧化沉淀溶液中的低价态金属离子,在适当的pH条件下,氧化生成的高价态金属离子会在溶液中形成氧化物(或羟基氧化物)沉淀,再通过过滤、烘干等工序后即可得到相应的金属氧化物粉体.金属氧化物制备过程中可能发生的反应如下:

臭氧氧化法制备金属氧化物既可避免因传统化学沉淀剂的加入而造成的二次污染,同时该工艺产物氧气清洁无污染,是一种新颖、高效的清洁制备工艺.
1.3 含氰废水处理
利用臭氧处理含氰废水过程中,臭氧首先与氰化物反应生成氰酸盐,然后生成的氰酸盐再与臭氧发生氧化水解反应,生成对环境友好的氮气和碳酸氢根离子,该过程中发生的反应为:
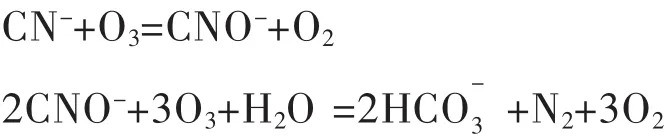
经臭氧处理后,废液中不含有害物质,无二次污染,不需要进一步处理,符合国家提倡清洁生产的要求,值得工业推广应用[4].
2 研究进展
2.1 难冶金属硫化矿浸出
2.1.1 难处理含金资源臭氧氧化预处理
金精矿经过氰化浸出,大部分易浸金都得以回收,但仍有少量金残留在尾渣中,氰化尾渣中大部分金以微细粒金的形式包裹于黄铁矿中,不能有效的与浸出剂反应,成为一种难处理的含金资源.
李登新等[5-6]开展了臭氧氧化浸出黄铁矿型金矿和氰化尾渣的系统研究:分别采用氯化铁、硫酸铁2种助氧化剂协同臭氧氧化浸出黄铁矿型金矿,在氯化铁联合氧化作用下,铁和硫的浸出率分别达到83.96%和72.56%;在硫酸铁联合氧化作用下,铁和硫的浸出率分别达到77%和66.7%,对最佳氧化条件下的氧化渣进行氰化浸金,金的浸提率达到96.5%.采用Mn2+催化臭氧氧化处理氰化尾渣[7],利用溶液中高价态锰和臭氧的协同氧化作用实现氧化浸出,铁的浸出率达到94.85%.Elorza-Rodríguez E等[8]对墨西哥的2种黄铁矿型金矿样品(A和B)在氰化浸金前采用臭氧氧化预处理,分别采用间接法(臭氧饱和水浸出3次)和直接法(直接将臭氧通入矿浆中)进行浸出.用间接法预处理,样品A在回收率方面没有明显变化,而用直接法预处理,在最大金和银回收率情况下,后续氰化时间从40 h降低到24 h;样品B用间接法使金回收率从53%提高到了88%,银回收率从26%提高到了78%.Pedroza F R C等[9-10]先后研究了利用臭氧氧化性预处理含金银黄铁矿原矿、尾矿和在稀硫酸体系中通过浸出煤炭中的黄铁矿、黄铜矿等含硫矿物进行煤炭脱硫,综合回收其中的金、银和铜等金属的工作.研究表明,臭氧预处理不仅提高了金、银和铜等金属提取率,而且可以减少低品位黄铜矿处理时间及后续工序氰化物的消耗量.
2.1.2方铅矿浸出
方铅矿是最重要的含铅矿物,其主体成分为PbS,利用方铅矿湿法提取金属铅主要方法是FeCl3浸出法,其优点是浸出速度比较快,但由于所用的氧化剂FeCl3价格较贵,对设备的腐蚀较强,因此该法较难在工业上推广应用[11].
湛雪辉等[12]探索了在盐酸溶液中,以臭氧和过氧化氢为氧化剂、三氯化铁为助浸剂联合浸出方铅矿精矿制备氯化铅的工艺.浸出过程中,方铅矿中的硫元素被过氧化氢、臭氧和氯化铁深度氧化成硫酸盐,同时生成氯化铅和相应的盐,而氯化铁中的Fe3+先被硫化铅还原为Fe2+,再被过氧化氢和臭氧氧化为Fe3+,起着助浸剂的作用.该工艺铅的浸出率达99.5%.此工艺直接制备得到合格的化工产品氯化铅,工艺流程短,环境污染少,产品附加值高.但工艺先在90°C高温下进行浸出,再通过冷却结晶得到产物,能耗较大,能源浪费严重.
2.1.3黄铜矿浸出
黄铜矿(CuFeS2)是自然界储量最大的铜矿,占世界铜矿资源的70%[13],黄铜矿的湿法浸出对铜工业具有重要意义.国内外很多研究者对黄铜矿的生物浸出、氯化浸出、氨浸、空气氧化浸出等进行了研究,但在常温常压下的浸出效果都不理想[14].
Havlik等[15]研究了硫酸体系下臭氧氧化浸出黄铜矿动力学.氧化浸出过程中,没有明显的单质硫层和任何其他产物层生成,表明臭氧氧化浸出有效地防止了钝化层的产生.
Carrillo等[16]在研究臭氧氧化浸出黄铜矿时发现浸出过程中会有大量的Fe2+形成,在臭氧较为充足的情况下,Fe2+会被臭氧氧化为Fe3+;在溶液pH值比较低,臭氧浓度比较高的条件下,溶液中Fe3+量会相对比较高,进而加速黄铜矿的浸出.
2.1.4黝铜矿浸出
原生黝铜矿(Cu12Sb4S13)结构稳定,分解困难,其浸出“钝化”问题会严重影响黝铜矿的进一步浸出.为克服浸出“钝化”,复杂硫化铜矿直接加压浸出时往往需要较高温度和较大压力(T>493 K,p>3.0 MPa).但是,高温高压条件对浸出设备及操作都提出了较苛刻的要求.如何克服黝铜矿浸出“钝化”,提高浸出速率,在较低温度及压力条件下实现高效浸出,已成为复杂硫化铜矿浸取研究的中心课题[17-18].
M.Ukasik等[19]研究了在盐酸体系中利用臭氧氧化浸出黝铜矿的方法.在最佳条件下,实验过程中铜所能达到的最大浸出率为60%,而锑氧化使部分锑形成沉淀,造成锑的最大浸出率只有20%左右.在浸出过程中可能发生的反应有:
2.1.5辉铜矿浸出
辉铜矿(Cu2S)是自然界中铜的主要矿物之一,其特点是含铜高,含硫、铁低,铁主要以磁铁矿或磁黄铁矿形式存在.近年来,利用湿法冶金技术处理辉铜矿的研究正在探索中.研究表明[20],辉铜矿第一个铜容易失去,随之转变为铜蓝,相对而言铜蓝比较稳定,受到H+攻击后晶格不容易破坏,浸出速率较慢.
Л·Н·克雷洛娃等[21]研究了联合臭氧与过氧化氢(Peroxone反应)形成氧化剂和硫酸铁浸出辉铜矿的新工艺.浸出过程中,辉铜矿按下式进行反应:
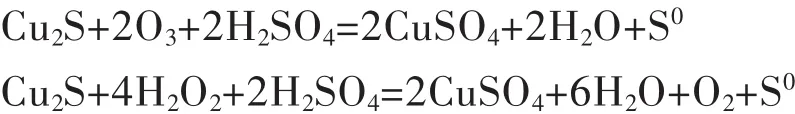
Havlík T[22]以臭氧为氧化剂研究了辉铜矿在稀硫酸溶液中的浸出过程.研究发现,当浸出5.5 h后,铜浸出率可达到97.32%.该工艺浸出温度较低,能量消耗少,臭氧用量少,环境友好.在浸出过程中硫酸消耗在硫化矿物和铁的氧化上,其消耗量部分可以依靠臭氧和过氧化氢氧化由硫化矿精矿浸出时形成的元素硫来补偿:
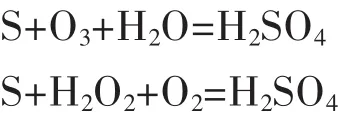
2.2 金属氧化物制备
2.2.1钴氧化物粉体材料制备
钴氧化物粉体材料是制备锂离子电池正极材料钴酸锂的主要原料,广泛应用于动力电池、磁性材料等领域.钴氧化物制备领域技术最成熟、应用最多的方法是沉淀-热分解法.但是,该法不仅消耗大量草酸铵,同时产生难以回收处理的含氨废水,直接排放不仅对环境造成严重危害,同时也是资源和能源的浪费[23].
田庆华等[24]采用臭氧氧化沉淀溶液中的Co2+,使其以羟基氧化钴(CoOOH)形式析出,在一定温度下经过煅烧后CoOOH可以转化为Co3O4.研究分析了浸出过程中发生的反应:
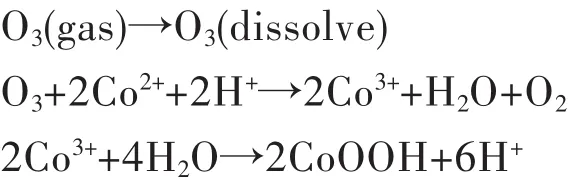
Bardé F 等[25]分别以 α-Co(OH)2为原料,通过直接暴露于臭氧中或向含钴溶液中通入臭氧的方法制备了γ-CoOOH和β-CoOOH,但是相比于以NaClO为氧化剂制备的γ-CoOOH电化学性能稍差.
Yuming Dong等[26]以醋酸钴和氨水为原料结合化学沉淀和水热法,以臭氧为氧化剂制备了Co3O4纳米粒子,并通过控制溶剂组合物 (乙醇与水的比例)或原料的浓度控制Co3O4纳米粒子的粒径大小.张爱民等[27]将化学沉淀和水热法结合,直接制备得到了Co3O4纳米颗粒和Co3O4纳米立方体.
Chung-Wei Kung等[28]研究了先利用电解沉积得到Co(OH)2然后利用紫外光/O3处理制备Co3O4的方法.紫外光/O3处理前后虽然钴氧化物薄膜的形态都是板状,但是经过臭氧处理后,钴氧化物薄膜由光滑变得粗糙,并且薄膜上出现微小的颗粒,处理后Co3O4的电化学性能相比处理前大大改善.
2.2.2羟基氧化镍制备
由于锌镍电池具有电压高、比能量大,比功率大和对环境友好等优点,近年来越来越引起国内外基础科学界和工业界的关注.羟基氧化镍(NiOOH)作为锌镍电池的正极活性物质,已经逐步成为电池材料领域一个新的研究热点.
Bardé F等[29]利用臭氧以 α-Ni(OH)2为原料在液相条件下制备了γ-NiOOH粉体.但是,由于水溶液中氢氧化钾等物质基本上不可避免,所以得到的产物不纯.Luis E.Calzad等[30]采用臭氧直接氧化硫酸镍(2 g/L)酸性溶液,并成功制备得到β-NiOOH粉体.研究发现,在室温条件下溶液pH值必须在6.8以上,臭氧氧化初始链反应才能发生.分析表明,反应实际上仍是先形成Ni(OH)2,然后Ni(OH)2再被臭氧氧化为β-NiOOH.该研究发现反应过程中需要过量的臭氧,分析认为这是由于臭氧会与溶液中碱性物质发生反应.反应如下:
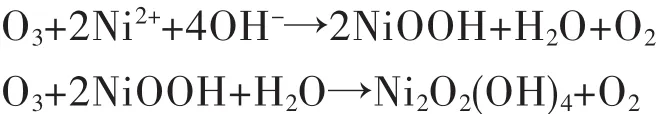
2.2.3银氧化物粉末制备
银氧化物(AgO)是一种灰黑色晶体或粉末,在蓄电池功率要求较高的军事、航空航天等高新技术领域和抗菌消毒领域具有广阔的应用前景.
沈文宁等[31]研究了臭氧氧化硝酸银制备高纯AgO超细粉末的新方法.制备过程中发生的反应如下:

臭氧氧化硝酸银制备AgO粉末工艺具有反应温度低,操作简单,原料气(空气)容易获得,反应副产物对环境无污染,AgO粉末制备所需费用低、纯度高的优点.
Waterhouse G I N 等[32]分别以 AgF(AgBF4)和CH3COOAg为原料,以臭氧为氧化剂,用液相方法制备了Ag3O4和Ag2O2等银氧化物,并证明了利用臭氧氧化制备Ag2O3是行不通的.制备过程中的反应可能为:

2.2.4二氧化锰粉末制备
二氧化锰是一种晶格结构比较复杂的氧化物,目前已知的20余种MnO2中,大多数是混合晶型,其氧化程度和水含量都是可变的[33].现有制备MnO2的方法主要有液相共沉淀法、低温固相法、热分解法、水热合成法和溶胶-凝胶法等.其中液相沉淀法以反应简单、快速、完全以及成本低廉等特点,成为粉体材料制备的研究热点.
Mtoski Sato等[34]公布了一种利用臭氧的氧化性将中性溶液中的二价锰离子氧化为二氧化锰沉淀的新发明.Susan J.Tewalt等[35]成功地在酸性溶液中利用臭氧从矿井排水中分离沉淀二氧化锰,综合回收废水中的锰资源.
Norihito Kijima等[36]研究了分别以硫酸、硝酸和盐酸为溶液体系,臭氧为氧化剂制备二氧化锰的方法.研究发现在硫酸体系中,得到的氧化产物为α-MnO2,而在硝酸体系及盐酸体系中得到的氧化产物为γ-MnO2,氧化产物中不含β-MnO2和斜方二氧化锰等晶型.
张爱民等[27]利用臭氧和可溶性二价锰盐溶液为原料,通过常温搅拌的方法,制备了β-MnO2纳米短棒;通过常温搅拌与水热法结合,分别在100~160℃和150~240℃下,制备了β-MnO2纳米针和β-MnO2纳米线.
2.3 含氰废水处理
金、银等金属提取过程会产生大量的含氰废水,如果未处理而直接排放,会对人类的健康造成巨大的威胁.目前,对于这类废水的处理大多采用碱性氯化法,但其存在处理成本高,易给环境带来二次污染等问题.酸化回收法适用于含氰废液的处理,主要是氰化物质量浓度较高的贫液处理,不适用于矿浆,且往往不能一次达标,需进行二次处理[4].
王长友等[37]研究了臭氧氧化法处理金矿氰化废水的工艺,在研究过程中,氰化物的去除率达97.9%,废水中氰化物的残余浓度低于0.5 mg/L.F.Barriga-Ordonez等[38]在研究臭氧氧化去除废水中氰化物过程中发现,该氧化反应的反应速率很大程度上取决于臭氧过量系数,臭氧过量系数越大,氰化物去除效率越高.Urszula Kepa等[39]分别探索了O3、H2O2和O3/H2O23种体系对去除废水中氰化物的效果.研究发现这3种体系中,高级氧化过程O3/H2O2对氰化物去除效率最高,而且氧化剂O3与H2O2的添加顺序也会影响氰化物去除效率,研究表明先加H2O2的效果比先加O3的效果更好.
3 存在的问题与前景展望
由于臭氧氧化处理含氰废水工艺流程短、相关操作及氧化反应机理比较简单,所以臭氧在含氰废水处理方面的研究及应用已经比较成熟,未来臭氧氧化法的研究重点将主要集中在难冶金属硫化物浸出和金属氧化物制备方面.经过多年的努力,臭氧在湿法冶金领域的研究取得了很大的进展.但是,臭氧氧化法发展至今仍然存在一些问题,主要体现在以下3个方面.
1)成本高.近年来,随着科学技术的进步,臭氧的生产成本逐渐降低,臭氧产生能力和臭氧在氧气中的浓度均得以提高.但是,在现有技术条件下,臭氧氧化法的投资和运营成本仍然很高.而且,考虑到臭氧本身是一种有毒气体,建立臭氧氧化系统需要采取严密的安全防护措施,这就提高了投资费用.成本问题一直是阻碍臭氧氧化法在湿法冶金领域得以推广应用的重要因素,国内尚无冶金企业将臭氧氧化法引入生产过程中的报道.
2)臭氧利用率低.现有的研究多为基础性的研究,反应体系一般比较小,而且臭氧作为一种气体,在溶液中的溶解度比较低,部分臭氧气体在溶液中还未参与反应就已经溢出溶液.此外,由于部分金属硫化物的浸出需要一定的温度,温度的升高不仅会降低臭氧在溶液中的溶解度,同时也会加速臭氧的分解,导致臭氧利用率比较低.
3)氧化机理不明确.尽管臭氧在金属硫化物浸出方面的研究已经取得很好的结果,在常温常压下即可实现各种难处理有色金属物料的浸出,对原料适应性很强,不仅有效地加快了硫化矿物的浸出速率,而且增大了金属的浸出率,但是,臭氧氧化浸出硫化矿物的机理还不是特别明确.在现有研究中各种金属硫化物臭氧氧化浸出的动力学特征和热力学数据还未完善,浸出过程中臭氧与硫化物的界面行为及产物硫的转变规律还需进一步探索,并建立一套明确的臭氧氧化浸出硫化矿物的机制系统.同时,臭氧在金属氧化物制备方面也只是报道了各种金属氧化物的制备,在制备过程中发生的晶型转变机理还未见报道.
综上所述,臭氧氧化法具有氧化效率高、选择性好、反应速度快等优点,且产物氧气不会引起二次污染,反应结束后可自行分解而没有残留,是一种新颖、高效的清洁冶金工艺.随着臭氧发生技术的进步,臭氧产生成本将会进一步降低,臭氧在湿法冶金领域中得以应用有望实现.而且臭氧的利用率与臭氧在溶液中的溶解度及停留时间密不可分,工业化应用时,由于反应体系的扩大,反应容器高度增大,压力增大,臭氧在溶液中溶解度增大,反应时间更长,同时改进曝气装置,改善臭氧在溶液中的分散性,臭氧的利用率将会极大的提高.目前,臭氧氧化法的研究重点是建立一套完善的、明确的氧化浸出机制系统,在原料方面,难冶含金、银等贵金属的复杂金属硫化物及低品位硫化矿将会是臭氧法研究的发展方向,金、银等贵金属的价格优势将会削弱臭氧成本所带来的影响,同时,加速该技术工业化的进程;在金属氧化物粉体材料方面,臭氧氧化机理研究及晶体转变的机制分析需要研究者进一步探索,为其他金属氧化物粉体制备提供方向和可行性的理论支持.
[1]Rischbieter E,Stein H,Schumpe A.Ozone solubilities in water and aqueous salt solutions[J].Journal of Chemical and Engineering Data,2000,45(2):338-340.
[2]Liu J L,Luo H J,Wei C H.Degradation of anthraquinone dyes by ozone[J].Transactions of Nonferrous Metals Society of China,2007,17(4):880-886.
[3]Bard A J,Parsons R,Jordan J.Standard potentials in aqueous solution[M].New York:Marcel Dekker Inc,1985:58-59.
[4]刘晓红,陈民友,徐克贤,等.臭氧氧化法处理尾矿浆中氰化物的研究[J].黄金,2005,26(6):51-53.
[5]钱方珺,李登新,李青翠.黄铁矿型难浸金精矿的试验研究[J].矿业工 程,2008,6(5):31-33.
[6]Li Q C,Li D X,Qian F J.Pre-oxidation of high-sulfur and higharsenic refractory gold concentrate by ozone and ferric ions in acidic media[J].Hydrometallurgy,2009,97(1):61-66.
[7]翟毅杰.臭氧催化氧化预处理氰化尾渣的研究[D].上海:东华大学环境科学与工程学院,2010.
[8]Elorza-Rodríguez E,Nava-Alonso F,Jara J.Treatment of pyritic matrix gold-silver refractory ores by ozonization-cyanidation[J].Minerals Engineering,2006,19(1):56-61.
[9]Carrillo Pedroza F R,Aguilar M J S,Martínez Luévanos A,et al.Ozonation pretreatment of gold-Silver pyritic minerals[J].Ozone:Science and Engineering,2007,29(4):307-313.
[10]Pedroza F R C,Aguilar M J S,Treviño T P,et al.Treatment of sulfide minerals by oxidative leaching with ozone[J].Mineral Processing and Extractive Metallurgy Review,2012,33(4):269-279.
[11]徐本军,覃文庆,邱冠周.方铅矿和软锰矿两矿法浸出工艺的研究[J].矿产保护与利用,2005(3):29-33.
[12]湛雪辉,李朝辉,湛含辉,等.臭氧-过氧化氢联合浸出方铅矿[J].中南大学学报(自然科学版),2012,43(5):1651-1655.
[13]Ashish P,Dastidar M G,Sreekrishnan T R.Bioleaching of heavy metals from sewage sludge:A review[J].Journal of Environmental Management,2009,90(8):2343-2353.
[14]蒋小辉,卢毅屏,冯其明,等.强氧化剂常温浸出黄铜矿及机理探讨[J]. 有色金属,2008,60(1):62-66.
[15]Havlik T,Dvorscikova J,Ivanova Z,et al.Sulphuric acid chalcopyrite leaching using ozone as oxidant[J].Metall,1999,53(1/2):57-60.
[16]Carrillo-Pedroza F R,Sánchez-Castillo M,Soria-Aguilar M,et al.Evaluation of acid leaching of low grade chalcopyrite using ozone by statistical analysis[J].Canadian Metallurgical Quarterly,2010,49(3):219-226.
[17]徐志峰,李强,王成彦.复杂硫化铜矿热活化-加压浸出工艺[J].中国有色金属学报,2010,20(12):2412-2418.
[18]徐志峰,李强,王成彦.复杂硫化铜精矿微波活化预处理-加压浸出工艺[J].过程工程学报,2010,10(2):256-262.
[19]Ukasik M,Havlik T.Effect of selected parameters on tetrahedrite leaching by ozone[J].Hydrometallurgy,2005,77(1):139-145.
[20]武彪,阮仁满,温建康,等.辉铜矿单矿物的氧化行为研究[J].有色金属,2008,60(3):58-61.
[21].Л·Н·克雷洛娃,李长根,崔洪山.采用新的药剂浸出有色金属硫化矿精矿[J].国外金属矿选矿,2008,45(7):41-42.
[22]Havlík T.Using ozone to intensify leaching of chalcocite[J].Transactions of the Technical University of Kosice,1992,2(2):231-237.
[23]Tian Q H,Guo X Y,Li Z H.Recovery of oxalic acid from motherliquor containing hydrochloric acid and cobalt by solvent extraction with P350[J].Transactions of Nonferrous Metals Society of China,2010,20:159-164.
[24]Tian Q H,Guo X Y,Yi Y,et al.Kinetics of oxidation-precipitation of cobalt(II)from solution by ozone[J].Transactions of Nonferrous Metals Society of China,2010,20:42-45.
[25]Bardé F,Palacin M R,Beaudoin B,et al.New approaches for synthesizing γIII-CoOOH by soft chemistry[J].Chemistry of materials,2004,16(2):299-306.
[26]Dong Y,He K,Yin L,et al.A facile route to controlled synthesis of Co3O4nanoparticles and their environmental catalytic properties[J].Nanotechnology,2007,18(43):435602.
[27]张爱民,董玉明,杨红晓,等.臭氧氧化制备 α-FeOOH、β-MnO2和Co3O4纳米材料的简便方法:中国,CN101037231A[P].2007-09-19.
[28]Kung C W,Chen H W,Lin C Y,et al.Synthesis of Co3O4nanosheets via electrodeposition followed by ozone treatment and their application to high-performance supercapacitors[J].Journal of Power Sources,2012,214:91-99.
[29]Bardé F,Palacin M R,Beaudoin B,et al.Ozonation:A unique route to prepare nickel oxyhydroxides.Synthesis optimization and reaction mechanism study[J].Chemistry of materials,2005,17(3):470-476.
[30]Luis E Calzado,Cesar O Gomez,James A Finch.Nickel recovered from solution by oxidation using ozone:Some physical properties[J].Minerals Engineering,2005,18(5):537-543.
[31]沈文宁,冯拉俊,孔珍珍.AgO超细粉末的制备及银的价态变化研究[J].稀有金属材料与工程,2011,40(11):1961-1965.
[32]Waterhouse G I N,Metson J B,Bowmaker G A.Synthesis,vibrational spectra and thermal stability of Ag3O4and related Ag7O8X salts[J].Polyhedron,2007,26(13):3310-3322.
[33]楼颖伟.纳米二氧化锰制备及电化学性能研究[D].杭州:浙江工业大学,2004.
[34]Mtoski Sato,Reston,VA(US);Eleanora I.Robbins,Vienna,VA(US).Recovery/removal of metallic elements from waster water using ozone:United States,US 6,485,696 B1[P].2002-11-26.
[35]Tewalt S J,Sato M,Dulong F T,et al.Use of ozone to remediate manganese from coal mine drainage waters[C]//Proceedings of National Meeting of the American Soc of Mining and Reclamation,2005:1166-1176.
[36]Norihito Kijima,Hiroyuki Yasuda,Toshio Sato,et al.Preparation and characterization of open Tunnel oxide a-MnO2precipitated by ozone oxidation[J].Journal of Solid State Chemistry,2001,159(1):94-102.
[37]王长友,祁金兵,张玲玲,等.臭氧氧化法处理金矿氰化废水的试验研究[J].辽宁化工,2004,33(8):446-447.
[38]Barriga-Ordonez F,Nava-Alonso F,Uribe-Salas A.Cyanide oxidation by ozone in a steady-state flow bubble column[J].Minerals Engineering,2006,19(2):117-122.
[39]Urszula Kepa,Ewa Stanczyk-Mazanek,Longina Stepniak.The use of the advanced oxidation process in the ozone+hydrogen peroxide system for the removal of cyanide from water[J].Desalination,2008,223(1):187-193.
