IL-32在急性心肌梗死中的表达及与其它细胞因子表达水平的相关性
2014-11-20陈家林余小平夏小杰
陈家林 余小平 夏小杰
深圳市第二人民医院,广东深圳 518036
急性心肌梗死是由冠状动脉急性、持续性缺血缺氧而引起的心肌坏死,有较高的发病率和死亡率,最根本的发病原因还是心血管疾病所造成[1]。患者多发生在冠状动脉粥样硬化狭窄的基础上,冠状动脉粥样斑块由于某些诱因而导致破裂,在破裂的斑块表面聚集着血中的血小板,之所以出现这种现象,是因为粥样斑块中的炎症因子在不断分泌。有研究表明,在心肌梗死的发生、发展过程、疾病的严重程度及预后的评估TNF-α和IFN-γ都发挥着重要的作用[2]。尤其最新发现的一种促炎症细胞因素IL-32,在对其他细胞因子调控时及炎症的发生发展中,可用IL-32进行调节[3]。目前,临床医学对IL-32的具体机制没有详细的阐明。该研究则是急性心肌梗死为切入点,分析L-32在此病的表达以及与 IL-1β、IL-6、IL-18、TNF-α 和 IL-10 等细胞因子表达水平的相关性,最终得出在急性心肌梗死中IL-32的作用[4],选取2012年12月—2013年11月该院急性心肌梗死的住院患者为研究对象,现报道如下。
1 资料与方法
1.1 一般资料
病例来自该院急性心肌梗死的住院患者,男24例,女12例,年龄47~72岁,均符合2001年中华医学会心血管病学分会“急性心肌梗死诊断和治疗指南”中的诊断标准[5],排除有糖尿病、恶性肿瘤、感染性疾病、肝肾功能不全等患者。对照组为30例体检健康者,男20例,女10例,年龄46~71岁。
1.2 标本的采集与测定
采集:急性心肌梗死组AMI组标本于患者发病的6 h内采集静脉血5 mL,静置,2 h内用3000 r/min离心10 min分离血清,于-40℃冷冻保存。对照组标本在早晨空腹时采集,处理方法同急性心肌梗死组。 测定:血清 IL-32、IL-1β、IL-6、IL-18、TNF-α、IL-10等含量采用酶联免疫法测定,采用酶联免疫吸附双抗体夹心法(深圳晶美生物工程有限公司)测定TNF-α及IL-18,采用ACL200型全自动血凝仪自动检测血浆中Fg浓度。操作均按试剂盒说明书进行。
1.3 统计方法
该次所有调查数据均采用软件SPSS20.0进行统计分析,两组 IL-32、IL-1β、IL-6、IL-18、TNF-α、IL-10 水平用()表示,比较采用t检验。
2 结果
两组血清 IL-32、IL-1β、IL-6、IL-18、TNF-α、IL-10 等水平比较,差异有统计学意义(P<0.05)。见表1。
表 1 两组 IL-32、IL-1β、IL-6、IL-18、TNF-α、IL-10 水平的比较()
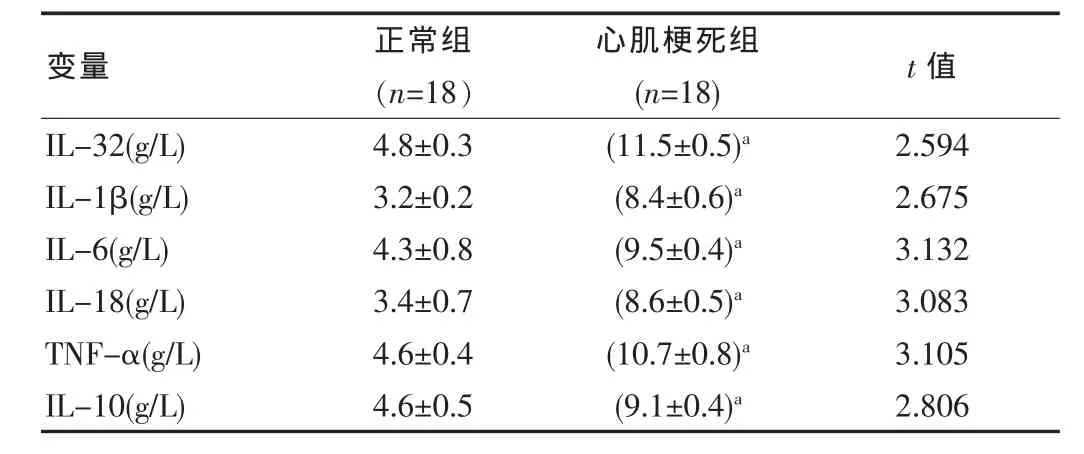
表 1 两组 IL-32、IL-1β、IL-6、IL-18、TNF-α、IL-10 水平的比较()
注:与正常组比较,aP<0.05。
变量 正常组(n=18)心肌梗死组(n=18) t值IL-32(g/L)IL-1β(g/L)IL-6(g/L)IL-18(g/L)TNF-α(g/L)IL-10(g/L)4.8±0.33.2±0.24.3±0.83.4±0.74.6±0.44.6±0.5(11.5±0.5)a(8.4±0.6)a(9.5±0.4)a(8.6±0.5)a(10.7±0.8)a(9.1±0.4)a 2.5942.6753.1323.0833.1052.806
3 讨论
炎症细胞因子指参与炎症反应的各种细胞因子,起主要作用的是 TNF-α、IL-1β、IL-6、TGF-β、IL-8 等,在心肌缺血缺氧损伤中也起着重要的作用[6]。如TNF-α是炎症反应过程中出现最早和最严重的炎性介质,激活淋巴细胞,增加血管内皮洗脑通透性。IL-6能诱导T细胞增殖和B细胞分化和产生抗体,是炎性反应的促发剂。IL-8能刺激中性粒细胞或T淋巴细胞的趋化,促进中性粒细胞脱颗粒,损伤内皮细胞。IL-32是新发现的炎症细胞因子,尤其在类风湿性关节炎及自身免疫疾病中,可激活NF-κB、P38/MAPK等信号通路,调整其他细胞因子的表达,使炎症反应的发展得以促进[7]。该研究则是急性心肌梗死为切入点,分析 L-32 在该病的表达以及与 IL-1β、IL-6、IL-18、TNF-α 和 IL-10等细胞因子表达水平的相关性,结果表明,心肌梗死组的心肌细胞在梗死后,只有IL-32表达水平升高,还有明显升高的则是抗炎症细胞因子IL-10和促炎症细胞因子 TNF-α、IL-1β、IL-6、TGF-β、IL-8。IL-32可在其他细胞因子缺氧的条件下上调或上述细胞因子的表达,导致心肌细胞损伤情况增重,也间接说明IL-32表达水平与个细胞因子表达水平的相关性呈正比。也有文献说明[8],在心肌缺氧过程中,IL-32的表达可能是通过NF-κB、P38/MAPK等信号通路上调上述。总之,IL-32基因及蛋白表达水平会在心肌细胞缺氧后明显升高,一定程度上会损伤其它细胞因子,进而加剧心肌细胞的损伤程度。
该研究结果表明,AMI发生时IL-32水平增高更为显著,IL-1β、IL-6、IL-18TNF-α 和IL-10的变化共同参与了 AMI的发生和发展,与国外的研究结果相一致[9],调节炎性细胞因子的释放均可能有利于减轻或阻止AMI的发生及严重程度,改善AMI患者的预后,抗炎因子治疗和调整机体免疫是治疗AMI的重要途径。
[1]Cai A,Zheng D,Dong Y,et al.Efficacy of Atorvastatin combined with adipose-derived mesenchymal stem cell transplantation on cardiac function in rats with acutemyocardial infarction[J].Acta Biochim Biophys Sin(Shanghai),2011,43:857-866.
[2]Wilensky RL,Hamamdzic D.The molecular basis of vulnerable plaque:potential therapeutic role for immunomodulation[J].Curr Opin Cardiol,2007,22:545-551.
[3]Kim SH,Han SY,Azam TA,et al.Interleukin-32:acytokine and inducer of TNF-α[J].Immunity,2005,22:131-142.
[4]Rosamond W,Flegal K,Furie K,et al.Heart disease and stroke statistics-2008 update:a report from the American Heart Association StatisticsCommittee and Stroke StatisticsSubcommittee[J].Circulation,2008,117:e25-e146.
[5]Goda C,Kanaji T,Kanaji S,et al.Involvement of IL-32 in activationinduced cell death in T cells[J].Int lmmunol,2006,18:233-240.
[6]Navarro—Lopez F.Genes and coronary heart disease[J].Rev Esp Cardol,2002,55:413-431.
[7]Carter AM.Inflammation,thrombosis and acute coronary syn-dromes[J].Diab Vasc DisRes,2005,2:113-12.
[8]Shoda H,Fujio K,Yamaguchi Y,et al.Interactions between IL-32 and tumor necrosis factor a1pha contribute to the exacerbation of immuneinflammatory diseases[J].Arthritis Res Ther,2006,8:R166.
[9]Dinarello CA,Kim SH.IL-32,a novel cytokine with a possible role in disease[J].Ann Rheum Dis,2006,65:61-64.
