二氯卡宾消耗臭氧的反应机理研究
2014-10-29石从云余加东胡磊刘兴重纪南南李德兵杨梦成
石从云,余加东,胡磊,刘兴重,纪南南,李德兵,杨梦成
(武汉科技大学化学工程与技术学院,湖北 武 汉430081)
0 引言
臭氧层的破坏给生物健康及生态平衡造成巨大的影响.用作制冷剂、发泡剂和溶剂的氟利昂、四氯化碳等卤代烃逸散到平流层中,在紫外线的作用下光解生成Cl、F原子和多卤代烃自由基等物质,这些物质会消耗臭氧[1-6].1CCl2是一种重要的光解产物[7-8],非常活泼,会与臭氧反应消耗臭氧.
当前人们对CCl2的关注主要见于光谱检测[9-11],及与水、氧气、氯气、乙炔、甲醛、一氧化氮和二氧化氮等小分子反应的动力学或机理等的研究[12-21].对于1CCl2与臭氧反应,前人仅报道了一种产物通道的形成过程[4-6].他们的研究表明反应第一步为无势垒络合生成四元环C—O—O—O中间体Cl2CO3,然后裂解生成产物Cl2CO+O2.我们认为反应物进攻很可能还有其他的方式,络合物经异构化和裂解过程可生成许多其他产物组合,且中间体Cl2CO3可以发生异构化生成别的中间体,然后裂解生成产物.文中用量子化学计算的方法对二氯卡宾和臭氧在单重态势能面上的反应进行了全面详细的研究,研究它们的反应机理,为控制CCl2消耗O3提供理论依据.
1 计算方法
采用密度泛函理论(DFT)在B3LYP/6-311G(d,p)计算水平上研究反应机理,优化反应过程中各驻点的几何结构,同时使用内禀反应坐标(IRC)在同一水平上计算确认过渡态与中间体之间的联系.利用在B3LYP/6-311G(d,p)水平上优化得到的各驻点结构,用 QCISD/6-311G(d,p)计算单点能的方法更精确地确定了各驻点的能量值.所有的计算都是用Gaussian03程序B.05版[22]完成的.
2 结果与讨论
通过计算得到1CCl2与1O3反应的8种产物通道,分别是P1(Cl2CO+1O2)、P2(CO2+ClClO)、P3(CO3+Cl2)、P4(CO+ClOOCl)、P5(CO2+ClOCl)、P6(v-CO2+ClOCl)、P7(ClCClO+1O2)、P8(COO+ClOCl).图1为优化得到的各驻点的几何结构.各驻点的振动分析表明,反应物、中间体和产物的力学常数矩阵本征值都为正,而过渡态有且仅有一个虚频.符号R表示反应物1CCl2+1O3,中间体用字母a、b、…、t表示,过渡态用符号“TSxy”表示,例如:TSac连接的是中间体a和c之间的过渡态.图2为单重势能面上可行反应路径的能级示意图,此图中的所有中间体和过渡态的能量都比反应物的低,在动力学上认为反应比较容易进行.图3为单重态势能面上不可行反应路径的能级示意图,此图中的每一条路径上都要越过比反应物能量高的过渡态,在动力学上认为反应难以进行.表1为在QCISD/6-311G(d,p)//B3LYP/6-311G(d,p)水平上计算所得的各驻点的总能量和相对能量.其中相对能量是以反应物的能量为零,其他各驻点的能量皆以反应物为基准,零点能校正值包含在单点能内.下面将详细描述反应路径,并从能量角度对反应路径进行分析以确定主要产物通道.
2.11CCl2+1O3的反应路径
2.1.11CCl2+1O3反应的可行路径 经图1中的可行路径共得到产物P1(Cl2CO+1O2)、P2(CO2+ClClO)、P3(CO3+Cl2)和P4(CO+ClOOCl).下面将对这4种产物通道的形成过程进行详细的描述.
1)反应物到产物P1(Cl2CO+1O2)
从反应物到产物P1(Cl2CO+1O2)有2条可行反应路径:Path P1(Ⅰ)、Path P1(Ⅱ).
Path P1(Ⅰ):R→a→TSap1→P1(Cl2CO+1O2)
首先,CCl2中的C原子进攻臭氧分子两端的氧原子(O1、O3)生成含有四元环(C—O1—O2—O3)结构且具有Cs对称(对称面为Cl1—Cl2—C—O2)的中间体a,然后中间体a中C—O3键(或C—O1键)与O1—O2键(或O2—O3键)断裂生成产物P1(Cl2CO+1O2).此过程经过的过渡态为TSap1,其虚频值为748.66i·cm-1,对应的振动模式为C—O3键(或C—O1键)与O1—O2键(或O2—O3键)的伸缩振动.该条路径与前人报道的一致[4-6].
Path P1(Ⅱ):R→a→TSac→c→TScd1→d1→TSd1g→g→TSgh→h→TShp1→P1(Cl2CO+1O2)
中间体a中的Cl1从C转移到O1原子上,同时O1—O2键断裂得到平面结构的中间体c.在这个过程中C—O1键键长由原来的0.139 1nm减小到0.130 3nm,C—O3键由0.139 1nm缩短为0.125 5nm,夹角O1—C—O3由94.65°增大到116.78°.然后O2从O3转移到C原子上,同时Cl1—O1绕C—O1键扭转64.84°生成中间体d1.紧接着C—O3键断裂,同时O3和O2靠近得到中间体g.此过程经过的过渡态为TSd1g,其虚频值为311.22i·cm-1,振动模式对应上述键的断裂和生成.中间体g形成后,Cl1从O1迁移到O2原子上便异构化为h.最后,中间体h中Cl1脱离O2并与C原子靠近成键,同时C—O2键断裂,生成产物P1(Cl2CO+1O2).
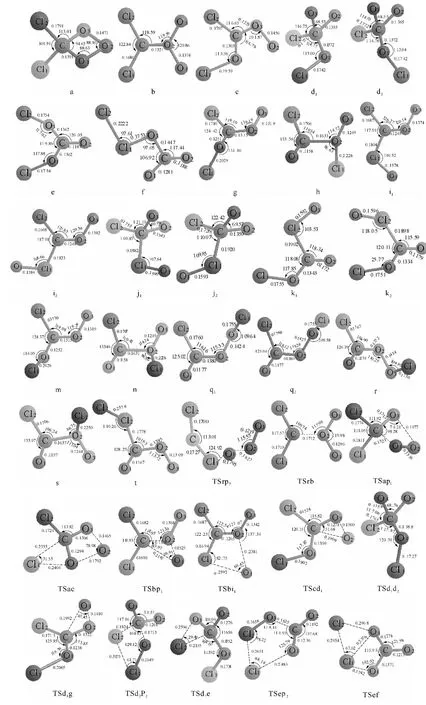
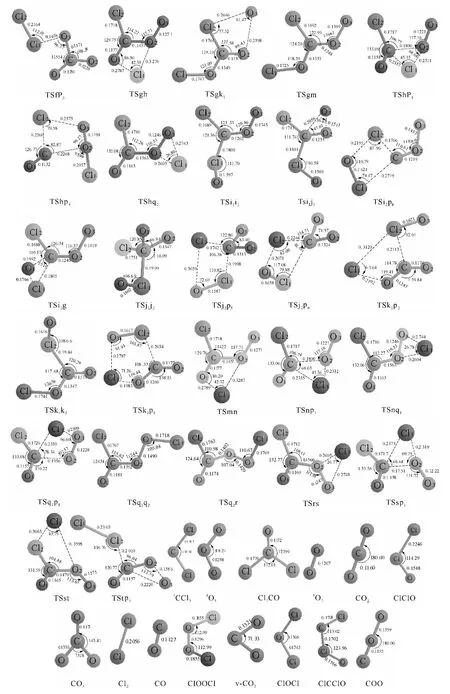
图1 1 CCl2+1 O3反应的反应物、中间体、过渡态和产物
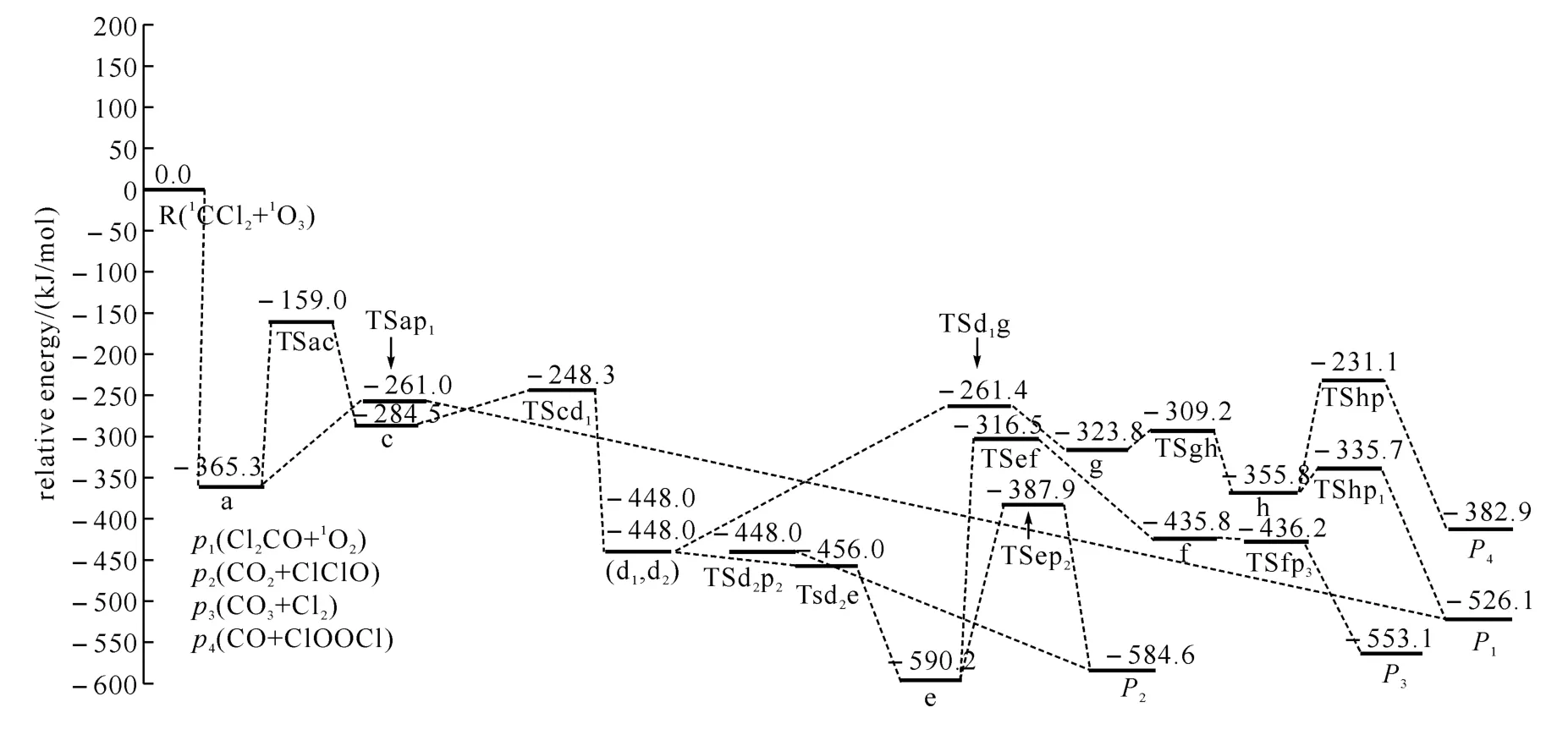
图2 1 CCl2+1 O3反应的可行路径在单重态势能面上的能级示意图
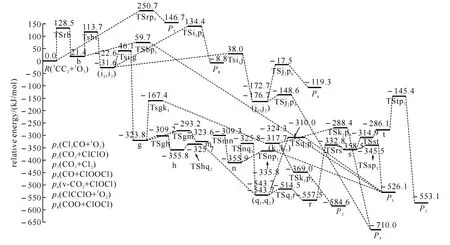
图3 1 CCl2+1 O3反应的不可行路径在单重态势能面上的能级示意图
2)反应物到产物P2(CO2+ClClO)
从反应物到产物P2(CO2+ClClO)也有2条可行反应路径:Path P2(Ⅰ)、Path P2(Ⅱ).
Path P2(Ⅰ):R→a→TSac→c→TScd1→d1→TSd1d2→d2→TSd2p2→P2(CO2+ClClO)
Path P2(Ⅰ)与Path P1(Ⅱ)的前几步(R→a→TSac→c→TScd1→d1)相同,前文中已经详细介绍了从反应物到中间体d1的转变过程,在这里就不重复介绍了.中间体d1中的O1—Cl1键绕着C—O1键旋转129.52°得到中间体d2.中间体d2与d1互为构象异构,为了简化路径,它们之间转换的过渡态在能级示意图中没有表示出来.中间体d2中的C—O1和C—Cl2键断裂,O2—C—O3由v型变为直线型,Cl2与Cl1靠近成键,从而生成CO2分子和ClClO自由基,即产物P2(CO2+ClClO).
Path P2(Ⅱ):R→a→TSac→c→TScd1→d1→TSd1d2→d2→TSd2e→e→TSep2→P2(CO2+ClClO)
中间体d2中Cl2从C转移到O3原子上生成具有C2对称的中间体e.在中间体e中,C、O2原子所在的直线为对称轴,C、O1、O2、O3原子处在同一个平面上,两个Cl原子处于该平面的两侧.然后,中间体e
中的Cl1转移到Cl2原子上,同时C—O3键断裂生成产物P2(CO2+ClClO).
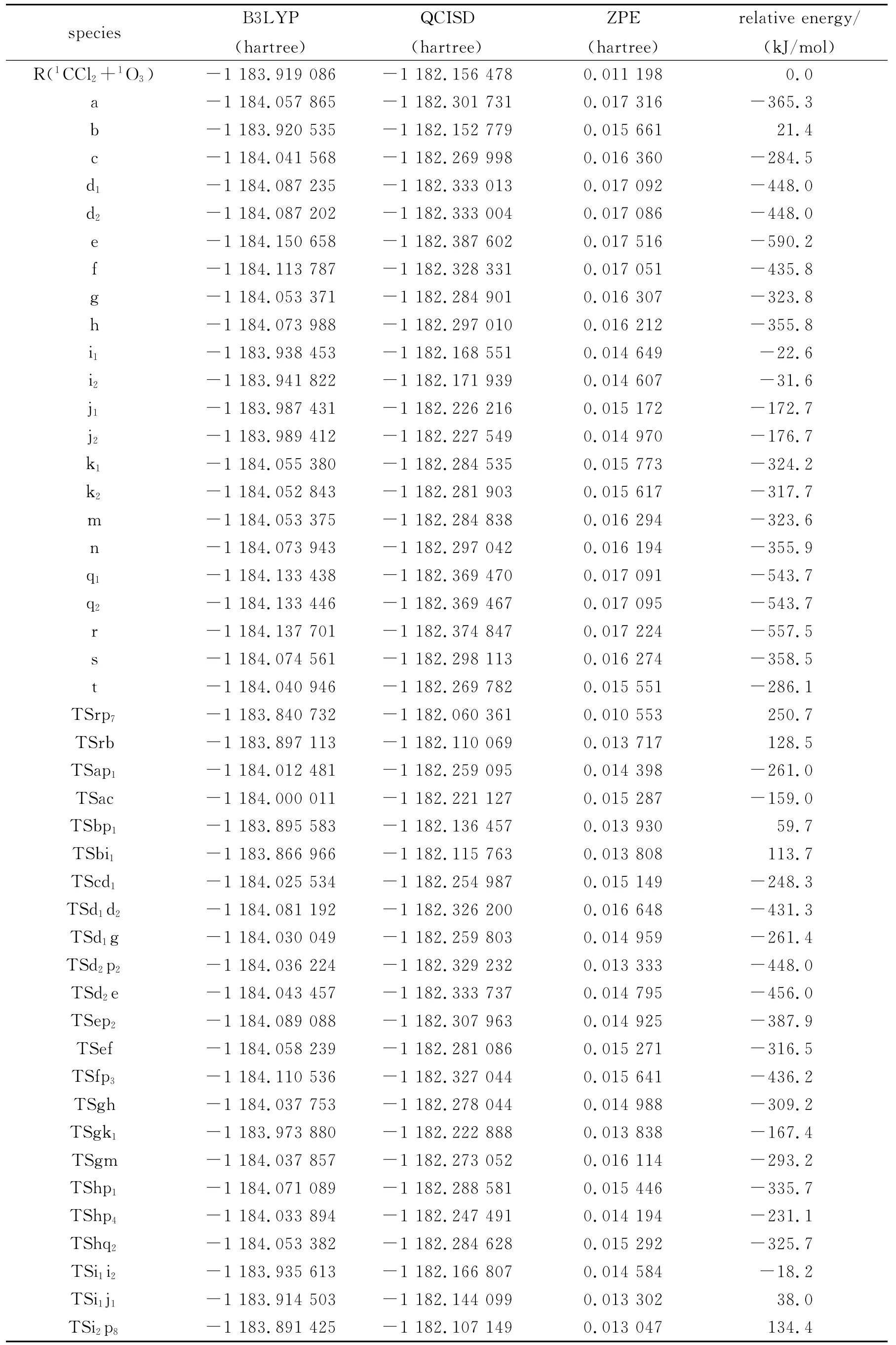
表1 1CCl2+1 O3反应的反应物、中间体、过渡态和产物在QCISD/6-311G(d,p)//B3LYP/6-311G(d,p)水平上计算所得能量值
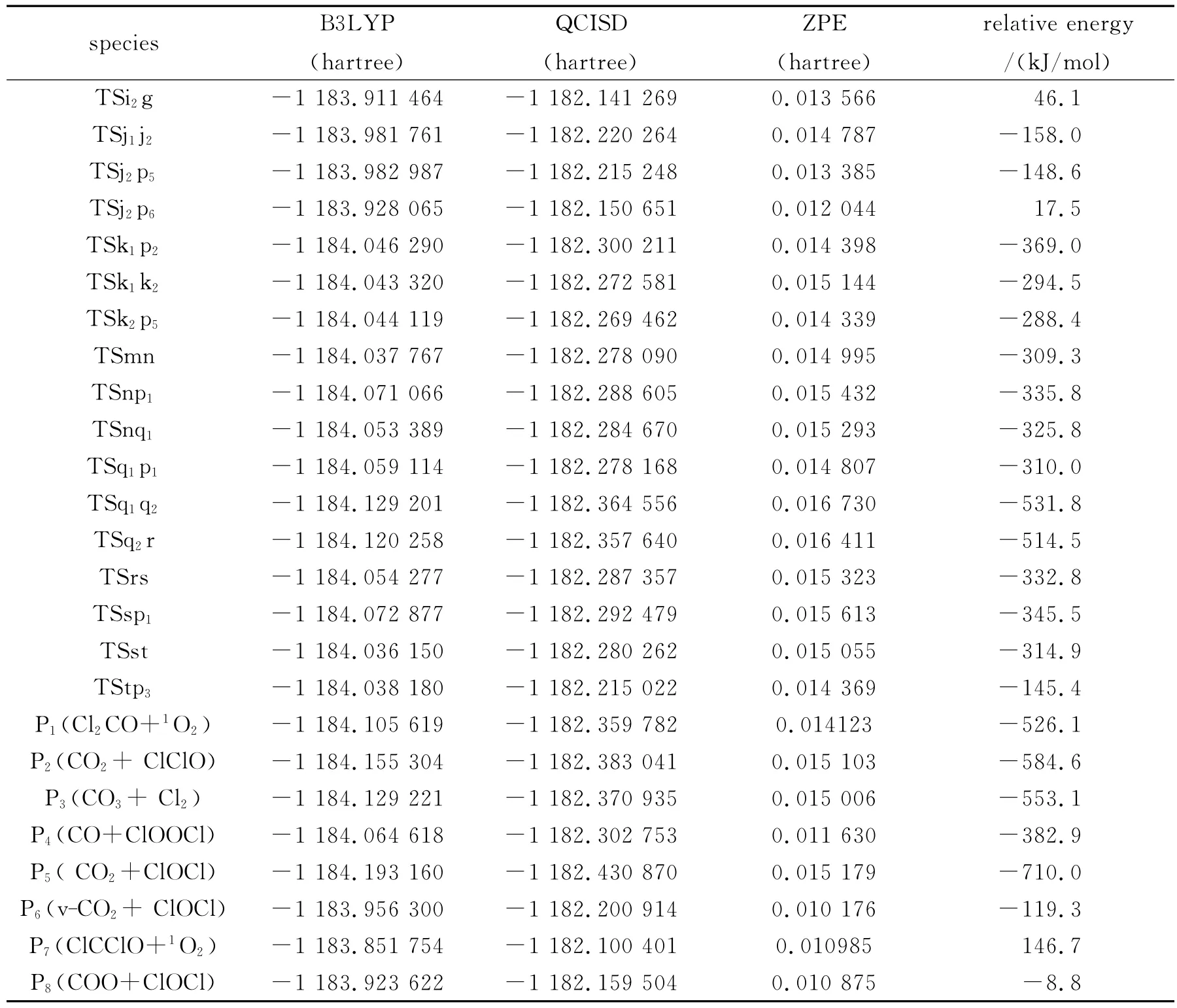
续表1
3)反应物到产物P3(CO3+Cl2)
从反应物到产物P3(CO3+Cl2)只有1条可行反应路径.
Path P3:R a→TSac→c→TScd1→d1→TSd1d2→d2→TSd2e→e→TSef→f→TSfp3→P3(CO3+Cl2)
中间体e中Cl1脱离O1并插入O3—Cl2键,使得O3—Cl2键断裂,同时形成O3—Cl1键和Cl1—Cl2键,从而得到平面构型的中间体f.此过程经过的过渡态为TSef,其虚频值为485.87i·cm-1,振动模式对应上述键的断裂和生成.最后O3—Cl1键断裂生成CO3自由基和Cl2分子,即产物P3(CO3+Cl2),且产物CO3具有C2v对称性.
4)反应物到产物P4(CO+ClOOCl)
从反应物到产物P4(CO+ClOOCl)也只有1条可行反应路径.
Path P4:R→a→TSac→c→TScd1→d1→TSd1g→g→TSgh→h→TShp4→P4(CO+ClOOCl)
中间体h中的Cl2原子从C原子迁移到O3原子上,同时C—O2键断裂,生成CO分子和ClOOCl自由基,即产物P4(CO+ClOOCl).
2.1.21CCl2+1O3反应的不可行路径 图2为不可行反应路径示意图.通过此图可把CCl2与O3反应的路径分为三类.第一,CCl2中的一个Cl原子进攻臭氧分子其中一端的氧原子O1,同时发生O1—O2键断裂,直接生成产物P7(ClCClO+1O2).此过程经过的过渡态TSrp7的相对能量非常高,为250.7kJ/mol,故可以认为此反应是很难进行的.第二,CCl2中的C原子进攻臭氧分子中间的氧原子O2,经过过渡态TSRb生成具有平面结构的中间体b,接着O1从O2迁移至C原子上,同时C—O2键断裂,得到产物P1(Cl2CO+1O2).此步的最高位过渡态TSRb的相对能量也比较高,为128.5kJ/mol,所以该反应也难发生.第三,中间体b生成后,O1从O2迁移到Cl1原子上经由TSbi1(113.7kJ/mol)生成平面构型的中间体i1,然后经过一系列的异构化和裂解生成产物P1、P2、P3、P5、P6和P8.由于过渡态TSrb和TSbi1都比反应物高出较多的能量,因而第三类反应也是不容易进行的,在此就不进行详细叙述了.
2.2 从能量的角度对1CCl2+1O3反应路径的分析 由于图2中的反应路径在动力学上可行,而图3中的反应路径在动力学上不可行,所以我们仅根据图2中的反应路径上的驻点的能量来判断反应的各产物通道的贡献.
在图2的6条可行路径中,Path P1(Ⅰ)过程比其他路径简单许多,而且Path P1(Ⅰ)上唯一的过渡态TSap1(-261.0kJ/mol)的能量低于其它路径最高位过渡态 TSac(-159.0kJ/mol)能量,所以Path P1(Ⅰ)的发生比其它路径容易得多.产物P1的生成除了这条最优路径外,还有一条过程复杂、过渡态能量较高的次要路径Path P1(Ⅱ),所以P1是最主要产物.
产物P2的形成也有两条路径,Path P2(Ⅰ)和Path P2(Ⅱ),它们的前几步(R→a→TSac→c→TScd1→d1→TSd1d2→d2)相同,后续过程不同.从中间体d2到产物的过程中,Path P2(Ⅰ)中的最高位过渡态的过渡态TSd2p2(-448.0kJ/mol)比Path P2(Ⅱ)中 TSep2(-387.9kJ/mol)能量低,故Path P2(Ⅰ)较Path P2(Ⅱ)容易进行.产物P3、P4的形成各只有一条可行路径:分别为Path P3和Path P4.因此通过比较Path P2(Ⅰ)、Path P3和Path P4的难易程度,就可得出P2、P3、P4对反应产物的贡献.对于Path P2(Ⅰ)和Path P3,从反应物到中间体d2的形成过程也一样,在后续过程中,Path P2(Ⅰ)的过渡态TSd2P2(-448.0kJ/mol)的能量明显小于Path P3上的过渡态 TSef(-316.5kJ/mol)的能量,因此Path P2(Ⅰ)比Path P3容易发生.用同样的方法我们也不难得出:Path P3较Path P4易进行.因而这三条路径发生由易到难的顺序为Path P2(Ⅰ)、Path P3、Path P4,相应的3个通道的产率依次为P2>P3>P4.产物P5、P6、P7和P8只能经动力学上不可行的路径生成,因而它们对1CCl2+1O3反应的产物体系几乎没有贡献.
3 结论
通过 QCISD/6-311G(d,p)//B3LYP/6-311G(d,p)水平的量子化学计算得到1CCl2+1O3反应的8种产物通道,即P1(Cl2CO+1O2)、P2(CO2+ClClO)、P3(CO3+Cl2)、P4(CO+ClOOCl)、P5(CO2+ClOCl)、P6(v-CO2+ClOCl)、P7(ClCClO+1O2)和 P8(COO+ClOCl),其中通道 P1(Cl2CO+1O2)是最主要产物通道,通道P2(CO2+ClClO)、P3(CO3+Cl2)、P4(CO+ClOOCl)的形成依次变难,而其他通道对反应产物的贡献可忽略.
[1]Imamura T,Chono H,Shibuya K,et al.Rate coefficient for the reaction of CCl3radicals with ozone[J].International Journal of Chemical Kinetics,2003,35(7):310-316.
[2]Ralph D G,Wayne R P.Reactions of the chlorodifluoromethyl radical formed in the photolysis of halogenocarbon+ozone mixtures[J].Journal of the Chemical Society,Faraday Transactions 2:Molecular and Chemical Physics,1982,78(11):1815-1823.
[3]李来才,周红平,田安民.F原子与臭氧反应机理的量子化学研究[J].物理化学学报,2002,18(1):59-61.
[4]殷平,陈先阳,张建强,等.单重态CCl2与 O3反应的热力学及动力学性质研究[J].化学学报,2000,58(11):1365-1368.
[5]胡海泉,刘成卜.单重态CCl2与O3反应机理的理论研究[J].化学学报,1999,57:29-33.
[6]李来才,周红平,王欣,等.单线态卡宾衍生物与臭氧反应机理的密度泛函理论研究[J].化学学报,2001,59(3):321-325.
[7]Tiee J J,Wampler F B,Rice W W.UV laser photochemistry of CCl4and CCl3F[J].Journal of Chemical Physics,1980,72(5):2925-2927.
[8]Sarah C A,Mary M K,Richard P J.Laser flash photolysis study of chlorocarbene[J].Journal of Chemical Physics,1996,100(47):18426-18430.
[9]Tiee J J,Wampler F B,Rice W W Jr.Laser-induced fluorescence excitation spectra of CCl2and CFCl radicals in the gas phase[J].Chemical Physics Letters,1979,65(3):425-428.
[10]Clouthier D J,Karolczak J.Pyrolysis jet spectroscopy:rotationally resolved electronic spectrum of dichlorocarbene[J].Journal of Physical Chemistry,1989,93(22):7542-7544.
[11]Choe J I,Tanner S R,Harmony M D.Laser-excitation spectrum and structure of CCl2in a free-jet expansion from a heated nozzle[J].Journal of Molecular Spectroscopy,1989,138(2):319-331.
[12]刘云珍,胡长进,裴林森,等.CCl2自由基与 H2O分子反应动力学研究[J].物理化学学报,2003,19(6):481-486.
[13]Josefredo R P Jr,Franca M A,Wagner B.Kinetics of the H2O+CCl2reaction in gas phase and in solution by an insertion mechanism[J].Chemical Physics Letters,1998,285:121-126.
[14]Alexander A S,Sofya A K,Eugene V S,et al.Kinetics of the reaction of the CCl2biradical with NO[J].Chemical Physics Letters,2003,381:766-770.
[15]Kostina S A,Shestov A A,Knyazev V D.Kinetics of the reaction of the CCl2biradical with molecular Chlorine[J].Journal of Physical Chemistry A,2003,107(48):10292-10295.
[16]李志锋,吕玲玲,康敬万.CCl2与CH2O插入反应机理及动力学特性的理论研究[J].化学学报,2007,65(11):1019-1026.
[17]Gao Y D,Ran Q,Chen Y,et al.State-state transitions for CCl2(X1A1,A1B1,a3B1)radical and collisional quenching of CCl2(A1B1and a3B1)by O2、N2、NO、N2O、NH3and various aminated molecules[J].International Journal of Chemical Kinetics,2002,34(6):351-356.
[18]Eskola A J,Golonka I,Rassanen M P,et al.Kinetics of the CCl2+Br2and CCl2+NO2reactions in the temperature range 266-365Kand reactivity of the CCl2biradical[J].Chemical Physics Letters,2008,460:401-405.
[19]Keating A E,Merrigan S R,Singleton D A,et al.Experimental proof of the non-least-motion cycloadditions of dichlorocarbene to alkenes:kinetic isotope effects and quantum mechanical transition states[J].Journal of the American Chemical Society,1999,121(16):3933-3938.
[20]Houk K N,Rondan N G,Mareda J.Are p-complexes intermediates in halocarbene cycloadditions?[J].Journal of the American Chemical Society,1984,106(15):4291-4293.
[21]林启君,冯大诚,戚传松.CX2(X== H,F,Cl)与甲基异丙基醚C—H键插入反应的理论研究[J].结构化学,2000,19(3):224-229.
[22]Frisch M J,Truks G W,Schlegel H B,et al.Gaussian 03,revision B 05[M].Pittsburgh:Gaussian Inc,2003.
