多类别肿瘤分类的特征基因选择方法研究
2014-09-22李小波彭司华
李小波,彭司华
(1.丽水学院工学院 计算机系,丽水323000;2.上海海洋大学水产与生命学院 生物技术系,上海201306)
近些年来,肿瘤的个性化医疗获得了很大的关注[1].由于肿瘤的异质性和患者之间的个体差异,如果简单采取同一方法治疗某一类肿瘤,就容易产生过度治疗或治疗不当的问题,因此,需要针对每一个患者的不同情况,采用个性化方式进行治疗.一个典型的例子是抗癌药物西妥昔单抗(Cetuximab,爱必妥),研究发现,西妥昔单抗对Kras基因野生型的结直肠癌患者效果显著,而对于Kras基因突变的患者则疗效欠佳,Kras基因的突变状态已经成为结直肠癌患者决定是否采取单抗靶向治疗的重要生物标志物[2].与此同时,肿瘤的个性化医疗也为肿瘤的分子分型和肿瘤生物标志物查找提出了紧迫的要求.
随着基因芯片技术的迅猛发展,利用基因表达谱对肿瘤进行分子分型,查找肿瘤标志物等工作取得了很大的进展[3].基因芯片技术能同时检测获取成千上万个基因的表达值,该技术为肿瘤研究开辟了一条高通量和系统性的研究途径,然而,基因芯片数据具有基因数量多(一般多于10 000)、样本数小(一般小于100)的特点,如何从中选取有效可靠的特征基因,则是基于基因芯片数据进行肿瘤分类的关键问题[4].通过基因选择,消除与肿瘤分类无关的噪声和冗余基因,获得精简的特征基因集,不仅可以减轻分类器的计算负担,还可以提高分类器的分类准确度.另一方面,所获得的特征基因集包含较少的基因数量,更便于后续的分子生物学实验验证,对于肿瘤标志物的查找和肿瘤发生发展分子机制的阐明具有实际意义.
常见的特征选择算法有3种[5]:过滤(Filter)法、缠绕(Wrapper)法和嵌入(Embedded)法.过滤法的选择结果与分类器无关,尽管使用过滤法选择基因方法简单快速,计算量小,但它也有几个不足之处:首先,过滤法忽视了与分类器的交互;其次,许多过滤法算法往往是一元的,并没有考虑到基因之间的相关性.缠绕法在某种程度上可以克服过滤法的上述问题.然而,该方法对计算的要求比较高,而且选择的基因集有较高的过拟合风险[6].嵌入法考虑到分类器的内部特征(比如支持向量机分类器里的支持向量),能与分类器较好地耦合,从而具有较高的精确度,但同时对分类器依赖性大,选择结果适应性差,需要检验其选择的基因对其他分类器的有效性[7].
目前在肿瘤的分类研究中,针对二分类问题(肿瘤类别数量为2,比如肿瘤样本与正常组织之间的分类)研究得较为透彻,且取得了较好的效果.而对于多类别肿瘤的分类问题(肿瘤类别或亚型的数量多于2),则缺乏深入的研究,并且在已有的研究结果中,可以看到准确度并不高,尤其当肿瘤类别越多时,分类准确度下降更快[8].肿瘤的发生发展和侵袭转移是多个阶段、多个基因调控、多条途径的过程[9],由此导致了肿瘤的异质性和肿瘤的多亚型.多类别肿瘤的分类问题,目前仍然是机器学习领域的一大挑战.
本文以肺癌的多类别基因表达谱数据为例,采用多步骤混合式特征基因选择策略[10],对多类别肿瘤的分类问题进行了研究.首先,采用7种常用的特征基因选择方法对基因表达谱中所有基因与分类的相关性进行排序,从中提取少量与肿瘤分类密切相关的基因子集,随后,对子集中的冗余基因进行过滤去除,得到分类准确度更高的精简基因集.实验结果证实了该方法的可行性和有效性.
1 特征基因选择方法
通过基因芯片检测到大量基因的表达值,其中多数基因与肿瘤相关性不大,对肿瘤的分类贡献也小,往往采用过滤法对所有基因的相关性进行排序,去除大量的低相关度基因,留下少量与肿瘤分类密切相关的基因.以下对本文涉及到的一些特征基因选择方法进行介绍.
1.1 基于相关性的特征选择方法
基于相关性的特征选择方法[11](Correlation-based Feature Selection,CFS)的核心思想是所选择的特征子集里的每个特征与每一个类高度相关,但相互间的相关度最低.评价特征子集的标准可以定义为

其中:Ms表示特征子集S的得分值,该子集含有k个特征表示子集内每个特征与类间的平均相关度表示子集内每个特征相互之间的平均相关度.
1.2 卡方选择法
卡方选择法[12](Chi-Squared,χ2)采用每个基因的卡方统计值(χ2)单独评价每个基因,首先将顺序或数字的特征属性值进行离散化,随后采用下面的公式计算每个基因的χ2值:

其中:m表示间隔数;n为类的数量;Aij表示第i间隔第j类的实际总模式数;Eij为Aij的理论频数.
1.3 信息增益选择法
信息增益选择法[13](Information Gain,IG)是基于熵的衡量方法,对于每个特征属性的计算公式如下:

其中:H(Class)为每个类的总熵;H(Class|Attribute)计算给定属性下每个类的条件熵.
1.4 信息增益率选择法
信息增益率选择法(Gain Ratio,GR)计算每个属性的信息增益率:
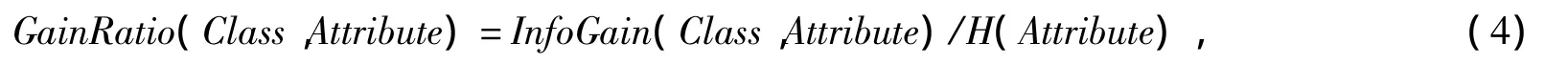
其中H(Attribute)表示每个属性的熵值.GR选择法用于衡量每个属性与类间的相对熵值.
1.5 对称不确定性选择法
对称不确定性选择法[11](Symmetrical Uncertainty,SU)计算每个属性的SU值:
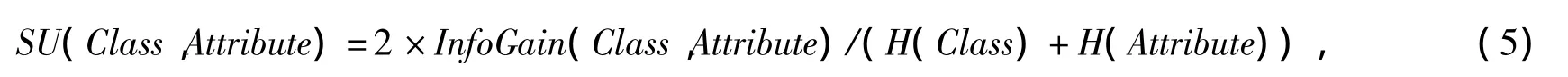
其中:H(Class)为每个类的总熵;H(Attribute)表示每个属性的熵值.SU选择法是针对信息增益选择法偏向选择具有较大信息增益值的特征而进行的调整.
1.6 ReliefF 选择法
在ReliefF选择法[14]中,良好的分类特征被定义为在同一类中具有相同的属性值,并在不同的类中具有不同的属性值.ReliefF法采用最近邻方法来计算每个属性的相关性得分.通过对实例的重复采样,根据最近的相同类和不同类的实例,评估每个属性的得分值.
1.7 基于支持向量机的递归特征基因消除法
Guyon等[7]提出了基于支持向量机的递归特征基因消除法(Support Vector Machine method based on Recursive Feature Elimination,SVM-RFE).SVM-RFE算法从原始基因集中逐个消除对分类器贡献最小的基因,基因对分类器贡献的重要性采用基因排序得分(gene ranking score)评估.基因排序得分定义为支持向量机权重向量w的平方和,w表示为

其中:xi为训练集中样本i的基因表达向量;yi∈[-1,+1]为样本i的类标签;αi可以从训练集计算得到.大多数样本的αi为零,αi为非零的向量为支持向量[7].
2 冗余基因的剔除和分类模型的评价
2.1 SSiCP 算法
通过上一节介绍的各种基因选择方法,可以去除大量与肿瘤分类无关的基因.设定一个阈值(比如设置基因子集中的基因数量),就能够获得一个与肿瘤分类存在高相关度的基因子集.然而,过滤法往往没有考虑到基因之间的相关性,在该子集中仍然存在一部分冗余基因.肿瘤特征基因选择方法的目标是获取一个基因数量尽可能少,包含样本分类信息尽可能全面的基因集,为此,文中采用了 Step-by-Step improvement of Classification Performance(SSiCP)算法[15].SSiCP算法采用一个评价函数,引导分类器逐步消除冗余基因.
该算法的关键步骤描述如下:
输入:训练集S1,该数据集包含特征基因数n1;
步骤1 训练特征基因数n的分类器,采用m-折(本文采用10-折)交叉检验法计算,得到分类器分类准确度k1;
步骤2 临时排除某个特征基因f,采用m-折(本文采用10-折)交叉检验计算,得到分类器分类准确度k2;
步骤3 假设k1≤k2,则消除特征基因f;假设k1>k2,则恢复特征基因f;
重复步骤1~3,直至分类器获得最高分类准确度;
输出:特征子集S2,特征基因数n2.
2.2 分类器的选择
支持向量机分类器具有较强的泛化能力,适合于具有高维数和小样本特征的肿瘤基因表达谱数据.文中采用Platt[16]提出的序贯极小优化(Sequential Minimum Optimization,SMO)算法是一个快速的支持向量机算法.在该算法中,将多类别分类问题化解为采用成对(pair-wise)分类的方式解决.
2.3 分类模型的评价
将肿瘤表达谱数据集按一定比例分为训练集和独立测试集,采用两种方法进行分类模型的评价:
(1)对于训练集,采用10-折交叉检验法(ten-fold cross validation)评价分类器,即将训练集中所有样本分为10等份,轮流将其中9份样本作为训练样本,剩余1份样本作为测试样本,不断重复测试,直至训练集内所有样本均被测试一次为止;
(2)对于独立测试集,以训练集中所有样本训练分类器,随后对独立测试集中的每个样本进行逐一分类测试.
机器学习算法中,过拟合(overfitting)是个必须要考虑的问题.给定假设空间H,一个假设h∈H,如果存在另外的假设h'∈H,使得在训练样本上h的错误率小于h',但在整个样本分布上h'的错误率小于h,那么就说假设h过拟合训练数据[17].因此,为了测试每个分类模型的过拟合风险,对于每个分类模型,首先在训练集中通过10-折交叉检验法获得一个分类准确度p,随后在独立测试集获得一个分类准确度p',如果p与p'的差别越小,说明该分类模型的抗过拟合性能越强.
3 结果
3.1 实验数据
采用 Bhattacharjee等[18]发表的肺癌数据集,数据从网站 http:∥www.pnas.org/content/98/24/13790/suppl/DC1下载.该数据集是个多类别肿瘤基因表达谱数据集,共包含203个样本,5个类别(4种肺癌亚型以及正常组织),其中腺癌(lung ADenocarcinoma,AD)127个,鳞状细胞癌(SQuamous cell lung carcinomas,SQ)21个,类癌(pulmonary CarcinOIDs,COID)20个,小细胞癌(Small-Cell Lung Carcinoma,SCLC)6个,以及正常肺组织样本(Normal Lung,NL)17个.其余的12例疑似肺外转移样本,未纳入本次实验.数据集中每个样本包含12 600个基因表达值.将整个数据样本按训练集相比独立测试集3∶1比例随机分配.肺癌数据集的结构如表1所示.
3.2 实验平台
首先对实验数据进行了预处理,分为两步:去除管家基因和标准化.去除管家基因后,数据集剩余12 533个基因表达值,对基因表达值进行标准化,使得每个样本的基因表达值均值为0,标准差为1.
本次实验中,在 WEKA(http:∥www.cs.waikato.ac.nz/ml/weka/)平台[19]上实现各种基因选择算法和分类模型.SMO分类算法有4种核函数 (Normalized Poly Kernel,Poly Kernel,RBF Kernel和String Kernel),我们选择采用多项式核函数(Poly Kernel),在训练支持向量机分类器的时候,调整最佳参数会是非常耗时的工作,因此我们采取参数固定的方式.其中由于数据已标准化,“FilterType”参数设定为“standardize training data”选项,并将惩罚参数C设为100.
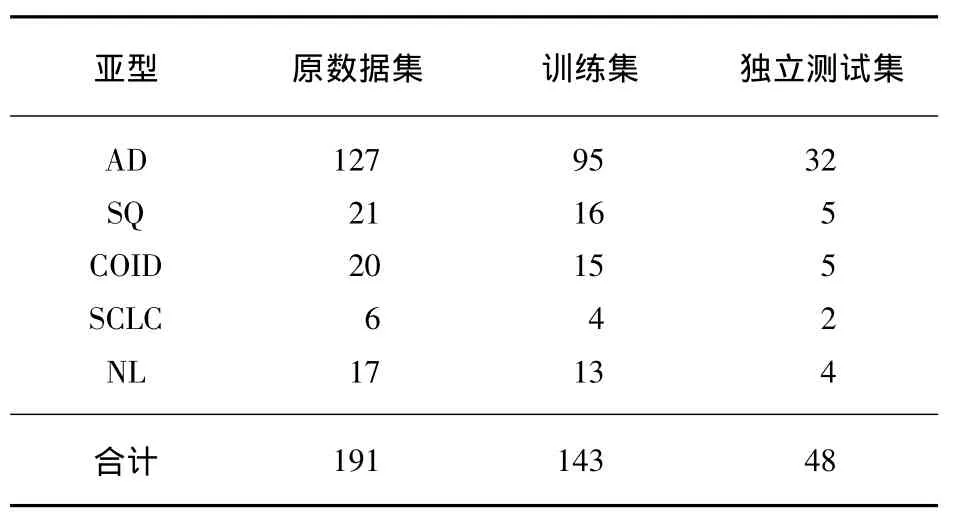
表1 肺癌数据集的样本分布Tab.1 Distribution of the samples in lung cancer data set
3.3 训练集的实验结果
在训练集的实验中,采用的是混合式基因选择策略.首先,采用 CFS、χ2、IG、GR、SU、ReliefF、SVM-RFE等基因选择方法去除与分类无关的基因,这些方法输出一个按得分由高至低排序的基因集.为了便于几种方法之间的比较,最后统一选择排序得分最高的200个基因,由此我们提取了与分类高度相关的基因子集.随后,考虑到这些基因之间可能存在强相关性,采用SSiCP算法进一步剔除具有强相关性的冗余基因.实验结果如图1所示.
由图1可知,SSiCP算法可以有效去除冗余基因.随着基因集基因数量的减少,分类预测精度经历了先上升而后下降的过程.SSiCP算法去除掉冗余基因后,非但不会减弱基因集的分类信息,反而进一步提高了预测精度,随着基因数量的减少,预测精度到达最高点并可继续维持,记录下预测精度最高且基因数量最少的基因子集,我们称之为“最精简”基因集.之后,随着基因数量的再次减少,预测精度则不断下降.
3.4 独立测试集的实验结果
采用各种基因选择方法获得的“最精简”基因集表达值,以训练集样本作为分类器的训练样本,建立分类模型,在独立测试集测试每个样本,记录每个独立测试集的错分样本,结果如表2所示.
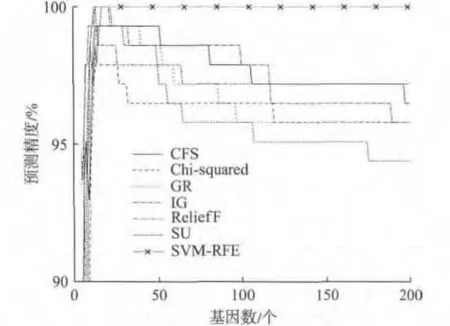
图1 SSiCP算法消减冗余基因过程中,7种不同基因选择方法的预测精度Fig.1 The prediction accuracies of the seven different gene selection methods when SSiCP algorithm was applied to eliminate redundant genes
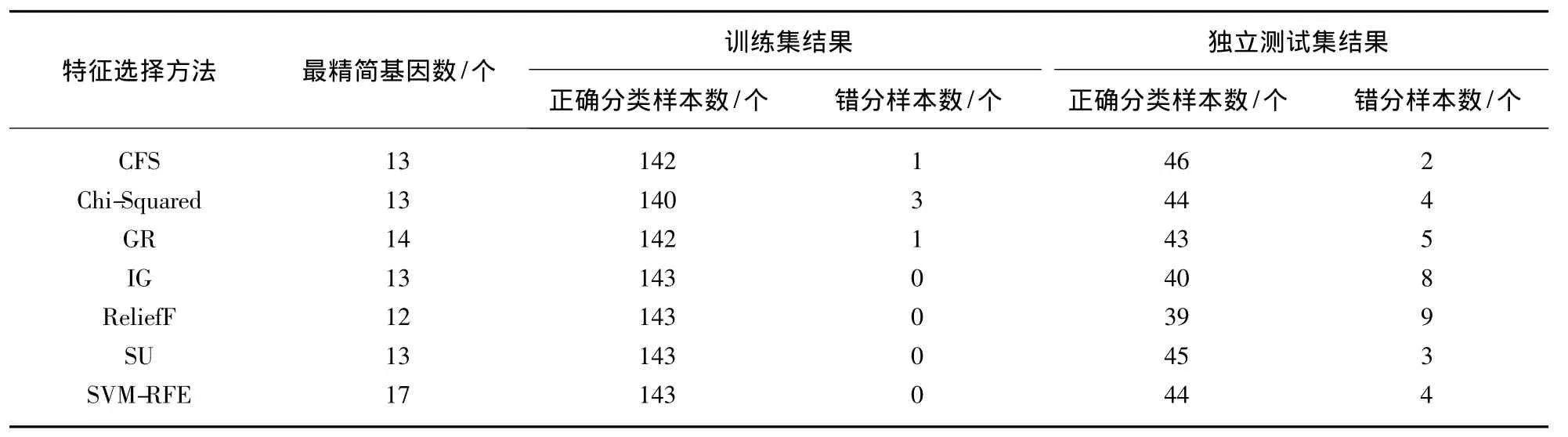
表2 在训练集和独立测试集中,7种特征选择方法的分类结果Tab.2 The classification results of the seven different gene selection methods in training and independent test data sets
表2综合给出了各种基因选择方法在训练集和独立测试集的性能.在训练集中,IG、ReliefF、SU和SVM-RFE等4种方法均获得100%的预测精度(见表2),其中ReliefF算法仅用了12个基因,表现最好.在独立测试集中,CFS算法的性能最佳,仅有2个错分样本.综合两个数据集的表现,SU算法在训练集的预测精度为100%,但在测试集的性能有所降低,有3个错分样本,精度为93.8%(45/48),在两个数据集的总体精度为98.4%(188/191).CFS算法在训练集有1个错分样本,在测试集有2个错分样本,总体精度和SU算法一致,为98.4%(188/191),然而,考虑到CFS算法在训练集的预测精度为99.3%(142/143),在测试集仅有2个错分样本,精度为95.8%(46/48),下降较少,其抗过拟合性能较强.两种方法的最精简基因数量均为13.因此,我们认为CFS算法在现有7种基因选择方法中有最佳分类性能.
3.5 最精简基因集
表3给出了采用CFS算法加SSiCP算法所获得最精简基因集的基因列表.其中至少4个基因,包括转录因子21(TCF21)基因[20],I型人T细胞白血病病毒结合蛋白3(TAX1BP3)基因[21],早幼粒细胞白血病锌指蛋白(ZBTB16或PLZF)基因[22],S100钙结合蛋白A8(S100A8)基因[23]等据文献报道与肺癌的发生发展密切相关.TCF21基因调节间充质细胞到上皮细胞的分化,在肺癌和头颈部鳞状细胞癌中显示出异常高的甲基化现象[24].Richards等[20]对105例非小细胞肺癌患者样本进行了研究,发现其中81%的样本存在TCF21基因启动子区甲基化现象,84%的样本表现出TCF21蛋白的表达下降.Wang等[21]在人类和小鼠肺癌细胞的研究表明,辐射诱导的细胞内TIP-1重新定位到质膜表面,可以抑制肿瘤细胞的增殖能力和集落形成能力,同时增强后续放射治疗的易感性.PLZF作为一种抑癌基因,其基因表达的失调在不同类型的实体肿瘤中有所报道.Wang等[22]探讨了其表达降低对非小细胞肺癌产生的影响.在154例配对非小细胞肺癌样本中,通过定量实时PCR实验发现,其中87.1%的样本PLZF表达下调了62.8%,并且35.6%的表达下调是由于PLZF启动子区甲基化引发的.在A549和LTEP肺癌细胞系中的实验发现PLZF的过表达能抑制细胞增殖和诱导凋亡.Su等[23]的研究采用了56例非小细胞肺癌和4例小细胞肺癌样本,免疫组织化学染色法和PCR法显示S100A8在肺癌组织中显著上调,发现其较高的表达与肺腺癌、肺部炎症和肺癌Ⅳ期病变的临床特征相关.

表3 CFS算法加SSiCP算法所获得的最精简基因集Tab.3 The minimum gene subset obtained by CFSplus SSiCPalgorithms

(续表)
采用 DAVID数据库(The Database for Annotation,Visualization and Integrated Discovery,DAVID)[25,26]分析表3最精简基因集的富集基因本体和通路,其中的功能注释图表工具(The Functional Annotation Chart tool)用于分析富集的注释.识别出3条富集的生物学过程(EASE score<0.05),包括DNA模板转录的负调控(GO:0045892~negative regulation of transcription,DNA-dependent),RNA代谢过程中的负调控(GO:0051253~negative regulation of RNA metabolic process)和转录负调控(GO:0016481~negative regulation of transcription),参与以上生物学过程的基因包括 TCF21,ZBTB16和BAZ2A等.
4 结语
由于肿瘤的异质性和肿瘤的多亚型特征,多类别肿瘤的分类问题是普遍存在的.随着高通量分子生物学技术的快速进展,采用基因表达谱等技术对肿瘤进行分子分型,已成为生物医学研究的热点,具有重要的理论意义和临床价值.文中重点研究了多类别肿瘤分类中的关键问题——特征基因选择方法.提出了混合式特征基因选择策略,并在肺癌的多类别基因表达谱数据集上进行了实验.首先用现有的7种特征选择算法各自提取了200个与分类高度相关的基因,随后采用SSiCP算法消除冗余基因,最终得到基因数量较少、分类准确度较高的最精简基因集.实验结果证实,SSiCP算法不但可以有效剔除冗余基因,并且能提高分类模型的准确度.比较了7种基因选择算法,发现CFS算法加SSiCP算法的混合式基因选择策略,获得了基因数量仅有13个的特征基因集,不但在训练集有较高的预测准确度,在独立测试集的准确度也比较高,有着较强的抗过拟合性能.采用CFS算法加SSiCP算法所获得最精简基因集中的部分基因,据文献报道,与肺癌的发生发展存在密切相关.文中所提出的混合式基因选择策略,可以作为多类别肿瘤分类中选择特征基因的有效方法.
[1]Chin L,Andersen J N,Futreal P A.Cancer genomics:From discovery science to personalized medicine[J].Nature Medicine,2011,17(3):297-303.
[2]Ong F S,Das K,Wang J,et al.Personalized medicine and pharmacogenetic biomarkers:Progress in molecular oncology testing[J].Expert Review of Molecular Diagnostics,2012,12(6):593-602.
[3]Golub T R,Slonim D K,Tamayo P,et al.Molecular classification of cancer:Class discovery and class prediction by gene expression monitoring[J].Science,1999,286(5439):531-537.
[4]Li X,Peng S,Chen J,et al.SVM-T-RFE:A novel gene selection algorithm for identifying metastasis-related genes in colorectal cancer using gene expression profiles[J].Biochemical and Biophysical Research Communications,2012,419(2):148-153.
[5]Saeys Y,Inza I,Larranaga P.A review of feature selection techniques in bioinformatics[J].Bioinformatics,2007,23(19):2507-2517.
[6]Leung Y,Hung Y.A multiple-filter-multiple-wrapper approach to gene selection and microarray data classification [J].IEEE/ACM Transactions on Computational Biology and Bioinformatics,2010,7(1):108-117.
[7]Guyon I,Weston J,Barnhill S,et al.Gene selection for cancer classification using support vector machines[J].Machine Learning,2002,46(1-3):389-422.
[8]Wang H,Zhang H,Dai Z,et al.TSG:A new algorithm for binary and multi-class cancer classification and informative genes selection [J].BMCMedical Genomics,2013,6(Suppl 1):S3.
[9]Li X B,Chen J,Lu B J,et al.-8p12-23 and+20q are predictors of subtypes and metastatic pathways in colorectal cancer:Construction of tree models using comparative genomic hybridization data[J].Omics-a Journal of Integrative Biology,2011,15(1-2):37-47.
[10]李小波.多步骤降维的肿瘤特征基因选择方法[J].复旦学报:自然科学版,2008,47(4):541-544.
[11]Mark A H.Correlation-based feature selection for discrete and numeric class machine learning[C]∥Proceedings of the Seventeenth International Conference on Machine Learning.San Francisco,CA,USA:Morgan Kaufmann Publishers Inc,2000:359-366.
[12]Huan L,Rudy S.Chi2:Feature selection and discretization of numeric attributes[C]∥Proceedings of the Seventh International Conference on Tools with Artificial Intelligence.Washington,D C,USA:IEEE Computer Society,1995:388-391.
[13]Wang Y,Tetko I V,Hall M A,et al.Gene selection from microarray data for cancer classification—a machine learning approach[J].Computational Biology and Chemistry,2005,29(1):37-46.
[14]Igor K.Estimating attributes:Analysis and extensions of RELIEF[C]∥Proceedings of the European conference on Machine Learning.Secaucus,NJ,USA:Springer-Verlag New York,Inc,1994:171-182.
[15]Peng S,Liu X,Yu J,et al.A new implementation of recursive feature elimination algorithm for gene selection from microarray data[C]∥Proceedings of the 2009 WRI World Congress on Computer Science and Information Engineering,Volume 03.Washington,D C,USA:IEEE Computer Society,2009:665-669.
[16]Platt JC.Fast training of support vector machines using sequential minimal optimization[M]∥Advances in kernel methods:Support vector learning.Cambridge,MA,USA:MIT Press,1999:185-208.
[17]Mitchell T.Machine Learning[M].Burr Ridge,IL,USA:McGraw Hill,1997.
[18]Bhattacharjee A,Richards W G,Staunton J,et al.Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses[J].Proceedings of the National Academy of Sciences of the United States of America,2001,98(24):13790-13795.
[19]Mark H,Eibe F,Geoffrey H,et al.The WEKA data mining software:An update [J].ACM SIGKDD Explorations Newsletter,2009,11(1):10-18.
[20]Richards K L,Zhang B,Sun M,et al.Methylation of the candidate biomarker TCF21 is very frequent across a spectrum of early-stage nonsmall cell lung cancers[J].Cancer,2011,117(3):606-617.
[21]Wang H,Yan H,Fu A,et al.TIP-1 translocation onto the cell plasma membrane is a molecular biomarker of tumor response to ionizing radiation[J].PloSOne,2010,5(8):e12051.
[22]Wang X,Wang L,Guo S,et al.Hypermethylation reduces expression of tumor-suppressor PLZF and regulates proliferation and apoptosis in non-small-cell lung cancers [J].The FASEB Journal,2013,27(10):4194-4203.
[23]Su Y J,Xu F,Yu J P,et al.Up-regulation of the expression of S100A8 and S100A9 in lung adenocarcinoma and its correlation with inflammation and other clinical features[J].Chinese Medical Journal(English),2010,123(16):2215-2220.
[24]Smith L T,Lin M,Brena RM,et al.Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer[J].Proceedings of the National Academy of Sciences of the United States of America,2006,103(4):982-987.
[25]Dennis G,Sherman B T,Hosack D A,et al.DAVID:Database for annotation,visualization,and integrated discovery[J].Genome Biology,2003,4(9):R60.
[26]Huang D W,Sherman B T,Lempicki R A.Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources[J].Nature Protocols,2009,4(1):44-57.
