中药网络药理学研究中的生物信息学方法
2014-09-12赵静方海洋张卫东
赵静,方海洋,张卫东
(1. 解放军后勤工程学院基础部,重庆 401311;2. 解放军第二军医大学药学院,上海 200433)
中药网络药理学研究中的生物信息学方法
赵静1*,方海洋1,张卫东2
(1. 解放军后勤工程学院基础部,重庆 401311;2. 解放军第二军医大学药学院,上海 200433)
为了更有效地治疗癌症、心血管疾病、免疫系统疾病等复杂疾病,基于分子网络的多靶点药物发现理念逐渐成为一种新的趋势,而中药整体、辨证、协同的用药观再一次引起了药物发现领域的极大兴趣。中药在治疗复杂慢性疾病方面有确切的疗效和较小的毒副作用。中药网络药理学从分子网络调控的水平上阐明中药的作用机制,为多靶点药物发现提供有益的启示和借鉴,并有可能从临床有效的中药反向开发现代多组分、多靶点新药。针对基于生物分子网络的中药药理学研究路线中的4个步骤,介绍近年来中药网络药理学研究中相关的生物信息学方法。
网络药理学;中药;疾病基因;药物靶点;信号通路;分子网络
中药(traditional Chinese medicine,TCM)治病历史悠久,在治疗复杂慢性疾病方面具有确切的药效和较小的毒副作用。然而,与西药相比,中药存在成分复杂、靶点和机制不清的问题。因此,在现代生物科学背景下阐明中药的作用机制仍是一个巨大的挑战。近年来,随着多靶点药物发现理念的兴起以及网络生物学研究的深入,网络药理学[1]、网络医学[2]等新的科学概念相继涌现出来,逐渐成为一个热门的研究模式,这对当代药物研究及新药发现产生了深入、全面且长远的影响。同时,将网络生物学的思想和方法应用于中药药理学研究,在分子网络调控的背景中研究中药的作用机制,也成为一种新的趋势,并产生了一系列新的数据、方法和成果。
中药网络药理学的核心问题之一,是评价中药的多个靶点的协同作用对疾病相关分子网络的综合影响问题。针对中药成分复杂、靶点不清的特点以及复杂疾病的网络调控特性,我们提出了基于生物分子网络的中药药理学研究路线[3],将基于分子网络的中药作用机制研究问题分解成4个步骤:1)识别中药的有效活性成分;2)识别各活性成分的作用靶标;3)识别中药所治疗疾病的相关基因并构建疾病网络;4)确定中药作用靶点所调控的信号通路和网络,评价中药对疾病网络的影响。本文将从生物信息学的角度,简要综述这4个方面相关的研究方法和进展。
1 中药有效活性成分信息的获取
尽管中药方剂通常含有成百上千种成分,真正起到治疗作用的是其中少数的活性成分。识别中药方剂中的有效活性成分,是了解整个方剂作用机制的基础。
中药研究及生物信息学领域的科学家在中药信息化建设方面进行了大量工作,构建了大量中药数据库。TCM数据库[4]、3D-MSDT[5]、TCMID[6]和 TCM Database@Taiwan数据库[7]等,均收录了中药及其主要化学成分的内容。其中TCM数据库根据《植物活性成分辞典》、《植物药有效成分手册》和《中药有效成分药理与应用》及相关文献编制,共收录相关的中药化学成分27 593种,包含每种成分的化学结构式、立体结构图、来源、药理作用、功效、临床应用、毒性、不良反应等信息[4]。3D-MSDT提供6 500种中草药有效成分的二维和三维结构以及其他各类相关信息,查询得到的分子可以直接在北京大学药物设计系统(PKUDDS)中进行三维结构的显示和分析[5]。TCMID收录了46 914种中药复方、8 159种中草药及25 210种中草药化学成分的相关信息[6]。TCM Database@Taiwan收录了453种中草药及它们所包含的2万多种化学成分的信息[7]。通过查询这些数据库,可以获得相关中药活性成分的信息。
2 药物作用靶标的识别
药物分子通过与特定的蛋白或核酸靶标相结合,调节其生物活性,从而发挥其治疗作用。因此识别中药有效活性成分所影响的分子,是认识中药方剂的作用机制必不可少的步骤。当前识别药物靶标的生物信息学方法主要有计算预测法和数据库查询法。
2.1 计算预测
计算预测方法采用计算机辅助药物设计技术,通过对药物分子和蛋白分子三维空间结构的模拟计算,预测药物的潜在靶标。当前流行的计算预测方法主要有反向分子对接、化学相似性搜索等。
反向分子对接(inverse docking)以小分子化合物为探针,在已知结构的候选靶点数据库内,搜寻可能与之结合的生物大分子,通过空间和能量匹配,识别可能形成的分子复合物,进而预测药物潜在的作用靶点[8]。2013年11月26日Nature杂志报道,纽约大学朗格医学中心的药理学家Timothy Cardozo近期在美国国立卫生研究院举办的“高风险-高回报”研讨会上,展现了他们所开展的人类有史以来最大规模的药物对接计算机模拟工作[9]。Cardozo研究团队从PubChem数据库和ChEMBL数据库中选择了大约60万种化合物分子,评价了这些分子与570种人类蛋白质的7 000多个结构“口袋”的结合能力的强弱。这项工作最终创建了一个名为Drugable的网站(网址:Drugable.com)并对公众开放。此数据库将极大地帮助对化合物靶标的识别。我们在研究黄芪的主要活性成分黄芪甲苷治疗心肌缺血的机制时,采用反向分子对接软件INVDOCK预测到黄芪甲苷的39个潜在作用靶点[10]。接着选择了预测的3个靶蛋白CN、ACE和JNK蛋白进行蛋白水平的实验验证,证实了黄芪甲苷确实对它们都具有抑制作用,但抑制的强度比对应的西药低。我们进一步开展了细胞水平的实验,比较黄芪甲苷和西药环孢菌素A对阿霉素诱导的心肌细胞损伤的修复作用。实验证实,黄芪甲苷与环孢菌素A均通过抑制乳酸脱氢酶(LDH)活性而产生对阿霉素诱导的心肌细胞损伤的修复作用,而黄芪甲苷的抑制作用低于环孢菌素A,但却显著高于蛋白水平的抑制作用。说明在细胞水平,黄芪甲苷可能通过作用于多个靶蛋白产生协同作用而使其作用显著增强。
化学相似性搜索(chemical similarity searching)[11]的基本假设是结构相似的分子倾向于与相似的靶标相结合。对于靶标未知的化合物,通过搜索与其结构相似而靶标已知的化合物,可以预测该化合物的靶标。2012年,美国诺华生物医学研究所的科学家用化学相似性搜索算法 (similarity ensemble approach,SEA),针对已知的73个药物副作用靶标蛋白,系统地研究了656种已获批准使用的药物的副作用靶标,他们预测到几百种以前未知的药物-蛋白相互作用,其中大约一半得到验证[12]。Zhang等[13]在研究乌头汤对类风湿关节炎的作用机制时,首先通过查询TCM Database@Taiwan数据库,获得乌头汤中165种化合物的化学结构,然后使用TTD(therapeutic targets database)数据库[14]所提供的药物相似性搜索工具,搜索到乌头汤中的化合物与153种药物有相似结构,从而预测了它的56个潜在作用靶点。李梢课题组[15]提出的靶标预测算法drugCIPHER将药物的化学相似性、治疗相似性相结合,在网络背景下预测药物靶标。利用其中的化学相似性预测模块,他们预测了清洛饮中235种活性成分分别对应的靶标。
2.2 数据库查询
基于前期大量的实验和计算方法产生的药物化学成分的靶标信息,近几年开发了大量靶标数据库,如DrugBank[16]、TTD、STITCH(Search Tool for Interactions of Chemicals)[17]以及PDTD[18]等;其中针对中药成分的靶标数据库有HIT (Herbal Ingredients' Targets Database)[19],TCM-PTD数据库(The Potential Target Database of Traditional Chinese Medicine,网址:http://tcm.zju.edu.cn/ptd/),TCM Database@Taiwan数据库等。这些数据库各有其侧重点,例如,DrugBank数据库收录了FDA批准的西药、实验阶段及研究阶段的西药的靶标信息;STITCH数据库收录了30万个小分子和1 133个物种中260万种蛋白质之间的相互作用信息,这些信息来自实验、数据库以及文献挖掘;HIT数据库包含了1 300种中药的586种成分与1 301种已知靶蛋白间的相互作用信息,这些信息是通过人工阅读文献收集的。通过查询这些数据库,可以获取中药有效成分的靶点信息,同时在研究中药时可以选取合适的西药作为对照。
笔者所在课题组在研究黄连解毒汤的抗类风湿机制时,针对前期实验识别的该复方的14种活性化合物,通过搜索HIT数据库获得91个靶标信息;同时从DrugBank数据库提取了FDA批准的4类抗类风湿关节炎西药32种,以及它们对应的51个靶标的信息,作为黄连解毒汤的对照组。我们发现,黄连解毒汤有5个靶蛋白与3类西药的靶蛋白一致,这在一定程度上解释了该方剂的抗类风湿作用[20]。
3 疾病相关基因的识别及疾病网络构建
直观看来,参与疾病发生发展过程的基因和蛋白应当是治疗此疾病的药靶,然而事实并不尽如此。Barabási课题组[21]通过构建和分析药靶与疾病基因间的关系网络发现,某些类别的疾病如内分泌、血液、心血管和精神类疾病的药靶偏好于疾病基因,而癌症、肌肉、骨骼、胃肠道和皮肤类疾病的药靶只有少部分是疾病基因。对于后一种情况,一般选择靶向与疾病基因相互作用的蛋白,或直接靶向该相互作用[22]。因此,识别疾病相关基因和构建疾病相关网络,是网络药理学研究的重要内容之一。
3.1 疾病相关基因信息获取
特定疾病相关基因的信息可通过数据库查询及文献挖掘的方法获取。目前已有许多数据库记录了疾病相关基因的信息,如在线人类孟德尔遗传数据库OMIM(Online Mendelian Inheritance in Man)[23]、遗传关联数据库GAD(Genetic Association Database)[24],GeneCard[25]等。OMIM数据库收录了迄今已知的人类孟德尔遗传性疾病、遗传决定的性状及其致病基因,并提供已知有关致病基因的连锁关系、染色体定位、组成结构和功能等信息。与孟德尔遗传性疾病不同,常见的复杂疾病,如心血管疾病、癌症、自身免疫性疾病、代谢性疾病等,是多因素的疾病,其特异的遗传基因并不清楚,几个基因的改变对特定个体的易感性也只有微小的贡献。GAD数据库则重点收录常见非遗传性复杂疾病以及与疾病关联的基因的信息,其信息主要来自文献报道。DisGeNET数据库[26]则收录了包括孟德尔遗传性疾病和复杂疾病的疾病-基因关联信息,该数据库是通过整合多个专家注释的数据库和文献挖掘的结果所构建的。通过查询这些数据库,可以获取疾病相关基因的信息。
3.2 疾病网络构建
与同一疾病相关的基因所编码的蛋白往往发生相互作用而形成疾病网络。然而,由于人类目前对复杂疾病的认识还远远不够,即目前已知的疾病基因的信息还只是很少的一部分,这些基因在人类全基因组背景网络中不一定连成一个连通的网络。由于在网络中,结点首先会影响它的直接邻居,最直观的方法是,将已知疾病基因连同它们在背景网络中的直接邻居,以及这些基因间的相互作用全部提取出来,构建成疾病网络。例如,Hwang等[27]研究哮喘病时,通过搜索OMIM数据库及分析GEO(Gene Expression Omnibus)数据库中哮喘病的基因芯片数据,获得606个哮喘病相关基因。将这些基因从由HPRD数据库[28]构建的人类全基因组蛋白相互作用网络中提取出来,获得的网络由297个互不相连的子网络构成,其中有269个孤立结点。而他们将这606个结点及它们在HPRD网络中的直接邻居提取出来,所构建的网络含2 438个结点,其中2 256个结点是互相连通的。相比而言,后一个网络更能反映哮喘病基因间的相互作用关系。但是,已知疾病基因的直接邻居,与疾病的相关程度并不一定是相同的。例如,基因A和B分别是1个和5个已知疾病基因的直接邻居,显然基因B与该疾病的相关程度更高;在全基因组背景网络中,若基因B和C都与5个已知疾病基因相连,但基因B的其他邻居个数显著比基因C的多,这2个基因与该疾病的关联程度也不同。因此,构建疾病网络需要对背景网络中的结点与已知疾病基因的关系作更深入的分析。
近年来,人们设计了一些算法,在已知疾病基因的基础上预测更多的疾病相关基因。其中,基于蛋白网络的算法的基本思想是,将已知疾病基因作为种子,在人类全基因组背景网络中,将结点按照它们在网络中与种子结点的邻近程度打分排序,排序靠前的基因则预测为疾病相关基因。这类算法可以用于疾病网络构建。即从背景网络中将已知疾病基因、预测的疾病基因,以及这些基因间的相互作用全部提取出来,构建成疾病网络。
不同的疾病基因预测算法从不同的角度量化网络中结点与种子结点的邻近程度。早期的算法如最近邻居法[29]、最短路径法[30]等采用局部网络邻近信息,后来的算法则应用全局网络邻近信息,如随机漫步法[31]、PageRank法[32]、k步马尔科夫法(k-step Markov method)[32]、网络传播法[33]、Katz-中心性法[34]等。Navlakha等[35]比较了不同算法,结果显示全局算法的结果显著优于局部算法。李梢课题组[36]提出的疾病基因预测算法CIPHER则将疾病间的相关性与网络结点的邻近性相结合,利用此算法,他们以OMIM中类风湿关节炎基因为种子,预测了更多类风湿关节炎相关的基因,从而构建了该疾病的网络。
为了理解免疫系统疾病脊柱性关节炎的病理机制,笔者所在课题组收集了GEO数据库中脊柱性关节炎的相关基因芯片数据并用统计方法找出显著表达的基因,同时收集了蛋白组学实验获得的该疾病状态下的显著表达蛋白,以及OMIM数据库中的脊柱性关节炎相关基因[37]。以这3个途径获得的168个疾病相关基因作为种子,利用笔者所在课题组提出的Katz-中心性算法[34],预测出更多的与该疾病相关的基因。所有这些基因编码的蛋白及它们间的相互作用,构成了与脊柱性关节炎相关的蛋白相互作用网络。该网络包含了大量自身免疫系统蛋白以及在骨髓中表达的蛋白,反映了该疾病是免疫系统紊乱导致的并损害骨骼关节系统的特性。
4 确定中药所调控的信号通路和网络、评价中药对疾病网络的影响
确定药物所调控的信号通路或子网络是药理学研究的核心问题。利用网络生物学的方法和技术,识别中药活性成分所调控的信号通路和影响的网络,并分析靶蛋白间的相互作用关系、靶蛋白在网络中所处的地位和起到的作用,可以帮助我们评价中药对疾病网络的影响,认识中药的作用机制。
4.1 药靶蛋白富集的信号通路识别
信号通路是执行重要的生物学任务和复杂的分子信息处理的、由分子间相互作用而形成的生物分子子系统或子网络。现有的信号通路数据库如KEGG[38]、Biocarta等收录了已知的信号通路、构成这些信号通路的基因及基因间相互作用的信息。如KEGG数据库收录了代谢、遗传信息处理、环境信息处理、细胞过程、有机体系统、人类疾病六大方面约500个信号通路。简单地将药靶蛋白直接映射到这些信号通路的基因集中,可以找到包含这些靶蛋白的通路。通常将药靶蛋白显著富集的通路认为是受药物调控的通路。
统计方法可以确定药靶蛋白富集的通路。超几何累计概率分布即是一种简单实用的方法。具体地讲,假设我们所研究的通路数据库中共有N个不同的基因,而某中药的药靶蛋白共有K个被映射到不同的通路上,那么,对于某个包含n个基因的通路,要衡量其上所映射到的k个药靶蛋白是否具有统计意义上的显著富集性,可用超几何累计概率分布所定义的P值来衡量。公式如下:
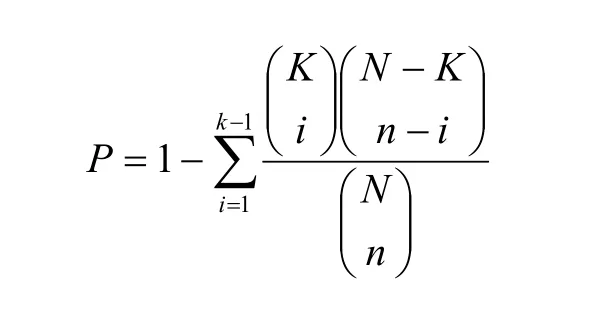
对给定的显著性水平a,若P值小于a,说明这k个药靶蛋白存在于这个含有n个基因的通路上是随机现象的概率小于a。因此可以将P值小于a的通路确定为药靶蛋白富集的通路。我们在研究黄连解毒汤对类风湿性关节炎的作用机制时[20],使用超几何累计分布方法找到了黄连解毒汤的靶标富集的32个信号通路,包括免疫相关信号通路、破骨细胞分化通路、细胞因子受体相互作用通路等。根据这些通路的功能,结合李梢课题组[39]对中医寒症、热症所对应的分子网络研究结果,可推断黄连解毒汤主要通过调节免疫和骨平衡,达到对热症型类风湿关节炎的治疗效果。Li等[40]使用缬草蜘蛛香的主要活性化合物异戊酸基二氢缬草素作用于卵巢癌A2780和OVCAR-3细胞系,发现该化合物对卵巢癌细胞的增长和繁殖起着剂量依赖的抑制作用,并通过基因芯片实验,识别到392和183个基因分别在该化合物作用后的A2780和OVCAR-3卵巢癌细胞系中差异表达,这些基因可看成卵巢癌细胞中受异戊酸基二氢缬草素所影响的基因。用超几何累计分布方法找出了该化合物调控的主要信号通路,其中有3个信号通路是在2种卵巢癌细胞系中都被显著调控的,其中包括p53信号通路。进一步的实验结果显示,缬草蜘蛛香通过激活p53的信号转导通路,诱导卵巢癌细胞凋亡和抑制卵巢癌细胞生长及其细胞周期进程。
基因集富集分析法(gene set enrichment analysis,GSEA)则定义了更有针对性的统计指标,包括富集分(ES)、标准化富集分(NES)、假阳性发现率(FDR)、标称P值(nominal P-value),用于识别某基因集所富集的通路[41]。GSEA软件包可免费下载使用,其中集成了常用通路数据库中的数据。
4.2 药物所影响的子网络的构建及治疗效应的评价
药物的靶标蛋白间相互作用的网络被认为是药物所影响的子网络。与构建疾病网络的情况类似,靶标蛋白在全基因组背景网络中不一定连在一起,因此需要建立一些合理的算法,确定网络中的结点和边。
最简单直观的方法,仍然是将靶标蛋白及其在背景网络中的直接邻居和相互作用全部提取出来,构建子网络。但这样的网络含有太多多余的结点和边,需要一定的简化。例如Huang等[42]构建青蒿素所影响的蛋白间的相互作用网络时,就先采用此方法得到一个初步的网络,然后再经过两步简化:1)删除连接蛋白之间的边;2)若2个靶标蛋白之间有多个连接蛋白,则删除连接度小的连接蛋白,最终获得一个简化的靶标蛋白间相互作用网络。笔者所在课题组在构建药靶间相互作用网络时,采用了图论中Steiner最小树算法,确定最少的靶标蛋白间的连接蛋白[10,43]。Antonov等[44]使用随机模拟的方法解决此问题,编写的软件PPI spider可自由下载使用。
如前所述,基于网络的疾病基因预测方法的原理,是量化网络中的结点与作为种子结点的疾病基因的邻近程度,从而可用于构建疾病网络。同理,当把药物的靶标蛋白取为种子结点时,这类方法同样可用于构建药物所影响的子网络。这类方法的优点在于,它可以用预测算法的所给的分值,来近似量化靶标蛋白对网络中其他蛋白的影响程度,从而得到药物对网络中各个蛋白影响程度的近似值。可以将影响分高于某个给定阈值的蛋白及其相互作用提取出来,构建药物所影响的子网络。
复方中药中不同成分间通过多靶标协同作用,形成集团作战的态势,进而发挥疗效。如何确定这种协同作用、量化评价药物作用于多靶标所达成的终极治疗效应,仍然是中药药理学研究中的难题。李梢课题组[45]提出的基于网络靶标的多成分协同作用识别法NIMS,将不同成分的靶标在疾病网络上的位置关系、与靶标对应的疾病谱的关系结合起来,确定存在协同作用的成分组合。笔者所在课题组在研究黄连解毒汤抗类风湿作用机制时,应用随机漫步算法,分别计算了类风湿关节炎的疾病基因和黄连解毒汤的靶标蛋白对全基因组网络中各蛋白的影响分,获得疾病影响向量和药物影响向量。用这2个向量的内积,分别量化评价黄连解毒汤的每个靶标、以及全体靶标对风湿关节炎疾病网络的总体影响水平,并与FDA批准的4种抗风湿西药以及随机对照进行了对比研究[20]。这些方法,代表了网络中药药理学研究从定性向定量研究的努力尝试。
5 展望
通过多种化学成分的组合用药,中药可以实现在远低于正常单一化学成分有效剂量下的同等药效。低剂量、多靶点、耐药性和毒副作用小这些用药特色,使得中药在治疗多基因调控的某些复杂疾病和需要长期用药的慢性疾病时,有着独特优势。当前,基于分子网络的“多靶点药物”发现的理念正逐渐成为一种新的趋势[1],并成为生物学、药物学和化学界的学术热点,且药物发现领域有一种向传统药物、民族药物获取资源和灵感的潮流[46]。近年来中药网络药理学研究证明,在细胞网络的背景下研究中药的主要成分及其靶点的作用,能帮助我们从本质上了解中药的整体、辨证、协调的用药观点。我们相信,在分子网络调控的水平上阐明中药的作用机制,将给多靶点药物发现提供有益的启示和借鉴,并有可能从临床有效的中药反向开发现代多组分新药,对中药现代化也将起着积极的推动作用。
[1]Hopkins A L. Network pharmacology: the next paradigm in drug discovery[J]. Nat Chem Biol, 2008, 4 (11): 682-690.
[2]Barabasi A L, Gulbahce N, Loscalzo J. Network medicine: a networkbased approach to human disease[J]. Nat Rev Genet, 2011, 12 (1): 56-68.
[3]Zhao J, Jiang P, Zhang W. Molecular networks for the study of TCM pharmacology [J]. Brief Bioinform, 2010, 11 (4): 417-430.
[4]任廷革, 刘晓峰, 高剑波. 中医药基础数据库系统介绍[J]. 中国中医药信息杂志, 2001, 8 (11): 90-91.
[5]Qiao X, Hou T, Zhang W, et al. A 3D structure database of components from Chinese traditional medicinal herbs [J]. J Chem Inf Comput Sci, 2002, 42 (3): 481-489.
[6]Xue R, Fang Z, Zhang M, et al. TCMID: traditional Chinese medicine integrative database for herb molecular mechanism analysis [J]. Nucleic Acids Res, 2013, 41 (D1): D1089-D1095.
[7]Chen C Y. TCM Database@ Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico [J]. PLoS One, 2011, 6 (1): e15939.
[8]Lybrand T P. Ligand-protein docking and rational drug design [J]. Curr Opin Struct Biol , 1995, 5 (2): 224-228.
[9]Reardon S. Project ranks billions of drug interactions [J]. Nature, 2013, 503: 449-450.
[10]Zhao J, Yang P, Li F, et al. Therapeutic effects of astragaloside IV on myocardial injuries: multi-target identification and network analysis [J]. PLoS One, 7 (9): e44938.
[11]Willett P, Barnard J M, Downs G M. Chemical similarity searching [J]. J Chem Inf Comput Sci, 1998, 38 (6): 983-996.
[12]Lounkine E, Keiser M, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets [J]. Nature, 2012, 486: 361-367.
[13]Zhang Y, Wang D, Tan S, et al. A systems biology-based investigation into the pharmacological mechanisms of Wu Tou Tang acting on rheumatoid arthritis by integrating network analysis [J]. Evid Based Complement Alternat Med, 2013, 2013: 548498.
[14]Chen X, Ji Z L, Chen Y Z. TTD: therapeutic target database [J]. Nucleic Acids Res, 2002, 30: 412-415.
[15]Zhao S, Li S. Network-based relating pharmacological and genomic spaces for drug target identification [J]. PLoS One, 2010, 5 (7): e11764.
[16]Wishart D S, Knox C, Guo A C, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration [J]. Nucleic Acids Res, 2006, 34: 668-672.
[17]Kuhn M, von Mering C, Campillos M, et al. STITCH: interaction networks of chemicals and proteins [J]. Nucleic Acids Res, 2008, 36 (Suppl 1): D684-D688.
[18]Gao Z, Li H, Zhang H, et al. PDTD: a web-accessible protein database for drug target identification [J]. BMC Bioinformatics, 2008, 9: 104.
[19]Ye H, Ye L, Kang H, et al. HIT: linking herbal active ingredients to targets [J]. Nucleic Acids Res, 2011, 39 (Suppl 1): D1055-D1059.
[20]Fang H, Wang Y, Yang T, et al. Bioinformatics analysis for the antirheumatic effects of Huang-Lian-Jie-Du-Tang from a network perspective [J]. Evid Based Complement Alternat Med, 2013, 2013: 245357.
[21]Yildirim M A, Goh K I, Cusick M E, et al. Drug-target network [J]. Nat Biotechnol, 2007, 25 (10): 1119-1126.
[22]Vassilev L, Vu B, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2 [J]. Science, 2004, 303: 844-848.
[23]Hamosh A, Scott A F, Amberger J S, et al. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders [J]. Nucleic Acids Res, 2005, 33 (Suppl 1): D514-D517.
[24]Becker K G., Barnes K C, Bright T J, et al. The genetic association database [J]. Nat Genet, 2004, 36: 431-432.
[25]Safran M, Dalah I, Alexander J, et al. GeneCards Version 3: the human gene integrator [J]. Database (Oxford), 2010: baq020.
[26]Bauer-Mehren A, Bundschus M, Rautschka M, et al. Gene-disease network analysis reveals functional modules in mendelian, complex and environmental diseases [J]. PLoS One, 2011, 6 (6): e20284.
[27]Hwang S, Son S W, Kim S C, et al. A protein interaction network associated with asthma [J]. J Theor Biol, 2008, 252 (4): 722-731.
[28]Keshava Prasad T S, Goel R, Kandasamy K, et al. Human protein reference database——2009 update[J]. Nucleic Acids Res, 2009, 37 (Suppl 1): D767-D772.
[29]Oti M, Snel B, Huynen M A, et al. Predicting disease genes using protein-protein interactions [J]. J Med Genet, 2006, 43 (8): 691-698.
[30]Krauthammer M, Kaufmann C A, Gilliam T C, et al. Molecular triangulation: bridging linkage and molecular-network information for identifying candidate genes in Alzheimer's disease [J]. Proc Natl Acad Sci USA, 2004, 101 (42): 15148-15153.
[31]Köhler S, Bauer S, Horn D, et al. Walking the interactome for prioritization of candidate disease genes [J]. Am J Hum Genet, 2008, 82 (4): 949-958.
[32]Chen J, Aronow B, Jegga A. Disease candidate gene identification and prioritization using protein interaction networks [J]. BMC Bioinformatics, 2009, 10 (1): 73.
[33]Vanunu O, Magger O, Ruppin E, et al. Associating genes and protein complexes with disease via network propagation [J]. PLoS Comput Biol, 2010, 6 (1): e1000641.
[34]Zhao J, Yang T H, Huang Y, et al. Ranking candidate disease genes from gene expression and protein interaction: a Katz-centrality based approach [J]. PLoS One, 2011, 6 (9): e24306.
[35]Navlakha S, Kingsford C. The power of protein interaction networks for associating genes with diseases [J]. Bioinformatics, 2010, 26 (8): 1057-1063.
[36]Wu X, Jiang R, Zhang M Q, et al. Network-based global inference of human disease genes [J]. Mol Syst Biol, 2008, 4: 189.
[37]Zhao J, Chen J, Yang T H, et al. Insights into the pathogenesis of axial spondyloarthropathy from network and pathway analysis [J]. BMC Syst Biol, 2012, 6 (Suppl 1): S4.
[38]Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes [J]. Nucleic Acids Res, 2000, 28 (1): 27-30.
[39]Li S, Zhang Z, Wu L, et al. Understanding ZHENG in traditional Chinese medicine in the context of neuro-endocrine-immune network [J]. IET Syst Biol, 2007, 1 (1): 51-60.
[40]Li X, Chen T, Lin S, et al. Valeriana jatamansi constituent IVHD-valtrate as a novel therapeutic agent to human ovarian cancer: in vitro and in vivo activities and mechanisms [J]. Curr Cancer Drug Targets, 2013, 13 (4): 472-483.
[41]Subramanian A, Tamayo P, Mootha V K, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles [J]. Proc Natl Acad Sci USA, 2005, 102 (43): 15545-15550.
[42]Huang C, Ba Q, Yue Q, et al. Artemisinin rewires the protein interaction network in cancer cells: network analysis, pathway identification, and target prediction [J]. Mol Biosyst, 2013, 9 (12): 3091-3100.
[43]Lu B, Zhao J, Xu L, et al. Identification of molecular target proteins in berberine-treated cervix adenocarcinoma HeLa cells by proteomic and bioinformatic analyses [J]. Phytother Res, 2011, 26 (5): 646-656.
[44]Antonov A V, Dietmann S, Rodchenkov I, et al. PPI spider: a tool for the interpretation of proteomics data in the context of protein–protein interaction networks [J]. Proteomics, 2009, 9 (10): 2740-2749.
[45]Li S, Zhang B, Zhang N. Network target for screening synergistic drug combinations with application to traditional Chinese medicine [J]. BMC Syst Biol, 2011, 5 (Suppl 1): S10.
[46]Verpoorte R, Crommelin D, Danhof M, et al. Commentary: "A systems view on the future of medicine: inspiration from Chinese medicine?"[J]. J Ethnopharmacol, 2009, 121 (3): 479-481.

[专家介绍] 赵静:女,1965年8月生。博士,解放军后勤工程学院教授,硕士生导师,应用数学学科带头人。先后于1985年、1988年在重庆大学应用数学系获应用数学学士、硕士学位,2008年在上海交通大学生命科学技术学院获生物信息学博士学位。1988年至今在解放军后勤工程学院基础部数学教研室工作。主要从事复杂网络理论在生物学、生物医学及中药药理学方面的应用研究。主持承担4项国家自然科学基金、2项863项目、多项省部级及军队级科研项目,获得重庆市自然科学二等奖一项、军队科技进步二等奖、三等奖各一项,获中国人民解放军院校育才奖银奖,荣立三等功一次。近年来在国内外学术期刊上发表相关论文30余篇,其中SCI论文20余篇。任多家国际学术期刊编委会成员和审稿人。
赵静教授课题组从2008年起与第二军医大学药学院张卫东教授实验室合作,用基于分子网络的系统生物学方法开展中药药理学的研究。开发了基于分子网络拓扑特性的算法,预测疾病致病基因、构建疾病相关的和药物干预的分子网络,用以分析复杂疾病的生物学过程及药物作用机制。应用所提出的研究路线和开发的算法,开展了一系列复杂疾病的病理以及特定的中药活性成分治疗相应的复杂疾病的机制研究,初步揭示了这些复杂疾病的分子网络特性,以及阐明所研究的中药活性成分的作用机制。在此领域已完成一项国家自然科学基金项目“基于网络生物学的药物分子作用机理研究”,相关研究成果发表在“Briefings in Bioinformatics”、“Journal of Proteomics Research”、“Journal of Ethnopharmacology”和“BMC Systems Biology”等知名期刊上。
Bioinformatics Approaches in Research on Network Pharmacology of Traditional Chinese Medicine
ZHAO Jing1, FANG Haiyang1, ZHANG Weidong2
(1. Department of Basic Science, Logistical Engineering University of PLA, Chongqing 401311, China; 2. Pharmacology School, No.2 Military Medical University, Shanghai 200433, China)
In order to treat complex diseases such as cancer, cardiovascular diseases and immune system disorders more effectively, the philosophy of molecular network-based multi-target drug discovery has gradually become a new trend. The holistic and synergetic medication mode of traditional Chinese medicine (TCM) has once again attracted a great interest in the field of drug discovery. TCM has shown exact therapeutic efficacy and low side effects in the treatment of chronic complex diseases. TCM network pharmacology seeks to clarify the action mode of TCM in the context of molecular networks. It may provide useful inspiration for drug discovery and develop the effective multi-component and multi-target drugs from clinically effective TCM along a reverse drug discovery path. Focusing on the four steps for network-based TCM pharmacology study, this review introduced the recent bioinformatics approaches applied in the research field of TCM network pharmacology.
network pharmacology; traditional Chinese medicine; disease gene; drug target; signaling pathway; molecular network
Q811.4
A
1001-5094(2014)02-0097-07
*接受日期:2013-12-21
项目资助:国家自然科学基金资助项目(No.61372194;No.10971227; No.81260672;No.81230090)
赵静,教授;
研究方向:系统生物学,中药药理学;
Tel:023-86730938;E-mail:zhaojanne@gmail.com
