大鼠星形胶质细胞Toll样受体表达谱
2014-09-12龚长银周爱玲
龚长银 周爱玲
(南通大学医学院病理生理学系,江苏 南通 226001)
近年来有研究表明,星形胶质细胞(AS)可以通过其表面的受体,如高级糖化终产物受体、清道夫受体等〔1,2〕及其介导的信号转导通路〔3〕而活化胞内信号转导机制,从而诱导前炎性递质等神经毒性物质的产生和释放,导致阿尔茨海默病(AD)神经炎症的发生〔4,5〕。然而,对AS Toll样受体(TLRs)的表达谱及其在AD炎症中的可能机制的研究未见诸多报道。TLRs是一种跨膜模式识别受体(PRRs),能够选择性地识别侵入机体的病原微生物所携带的病原相关分子模式(PAMPs),在病原识别、启动和指导机体的免疫应答中发挥极其重要的作用〔6~8〕。本文旨在探讨AS TLRs表达谱系。
1 材料与方法
1.1实验动物 新生1~3 d SD大鼠,雌雄不限,由南通大学实验动物中心提供,许可证号:SYXK(苏)2002-0022。
1.2主要试剂及仪器 DMEM/F12、胰蛋白酶、L-谷氨酰胺、胎牛血清(杭州四季青)、多聚赖氨酸(Sigma)、实时PCR试剂、TLRs引物(上海睿安生物科技有限公司,见表1)、琼脂糖、Trizol(Invitrogen)、mouse anti-GFAP monoclonal antibody及Rabbit anti-TLR4 polyclonal antibody(Santa Cruz)、Hoechst 33342(Dojindo)。BS110S型电子天平(北京赛多利斯天平公司)、Microfuge 22R台式冷冻离心机(Beckman)、-80℃超低温冰箱(SANYO)、PHS-3C数字式酸度计(江苏电分析仪器厂)、PTC-100 PCR仪(Bio-rad)、SX-300凝胶成像系统(上海四星生物技术有限公司)、小源凝胶图像分析系统(上海小源科技有限公司)、移液器(Eppendorf)、IX51倒置荧光相差显微镜(SANYO)。
1.3AS的分离及培养(在无菌条件下操作) 取新生1~3 d的SD大鼠,75%乙醇浸泡消毒5~10 min,用眼科剪断头,在无菌条件下用眼科剪小心剪开头骨,剥离颅骨后小心取出双侧大脑皮层,置于盛有预冷D-Hank′s液中的培养皿中,轻轻地剥离脑膜和血管。然后,将大脑皮层移至另一个盛有预冷D-Hank′s液的培养皿中,如此洗2~3遍,以尽可能的去除脑膜和血管等。最后,将大脑皮层置于盛有DMEM/F12基础培养基的培养皿中,用眼科剪将大脑皮层剪成糜状,用巴斯德滴管转移至离心管中,加入等体积的0.25%胰蛋白酶消化液(终浓度为0.125%),37℃下消化10 min,每5分钟晃动1次。然后,用含血清的培养基终止消化,轻轻吹打分散细胞,用不锈钢滤网过滤,1 000 r/min离心5 min。弃上清,加入含血清的培养基,重新混悬沉淀成单细胞悬液,进行细胞计数。调整细胞浓度,按(1~2)×106/ml接种于未包被多聚赖氨酸的培养瓶中,于37℃、5% CO2培养箱中差速黏附处理30~60 min后,轻轻翻转培养瓶,取细胞悬液接种于经多聚赖氨酸包被的培养瓶中继续培养。每2~3天换液1次。培养9~14 d待细胞基本融合成单层并铺满瓶底后,进行消化传代培养。
1.4AS的传代纯化 从培养箱中取出培养瓶,充分振荡,用巴斯德吸管吸除培养液后,再用D-Hank′s液洗两遍。用0.125%胰蛋白酶消化,并在倒置显微镜下观察细胞消化情况,待细胞变圆,大约有2/3的细胞漂浮在液体中时立即终止消化,防止消化过度而影响AS的活力。用吸管反复吹打培养瓶的底部,尽可能地将贴壁的细胞吹打下来。用培养基调整细胞密度,最后按照(1~2)×106/ml接种至预先经多聚赖氨酸处理的培养瓶中继续培养。如此,待细胞传至3~4代后,进行AS纯度鉴定,达到要求后用于后续实验。
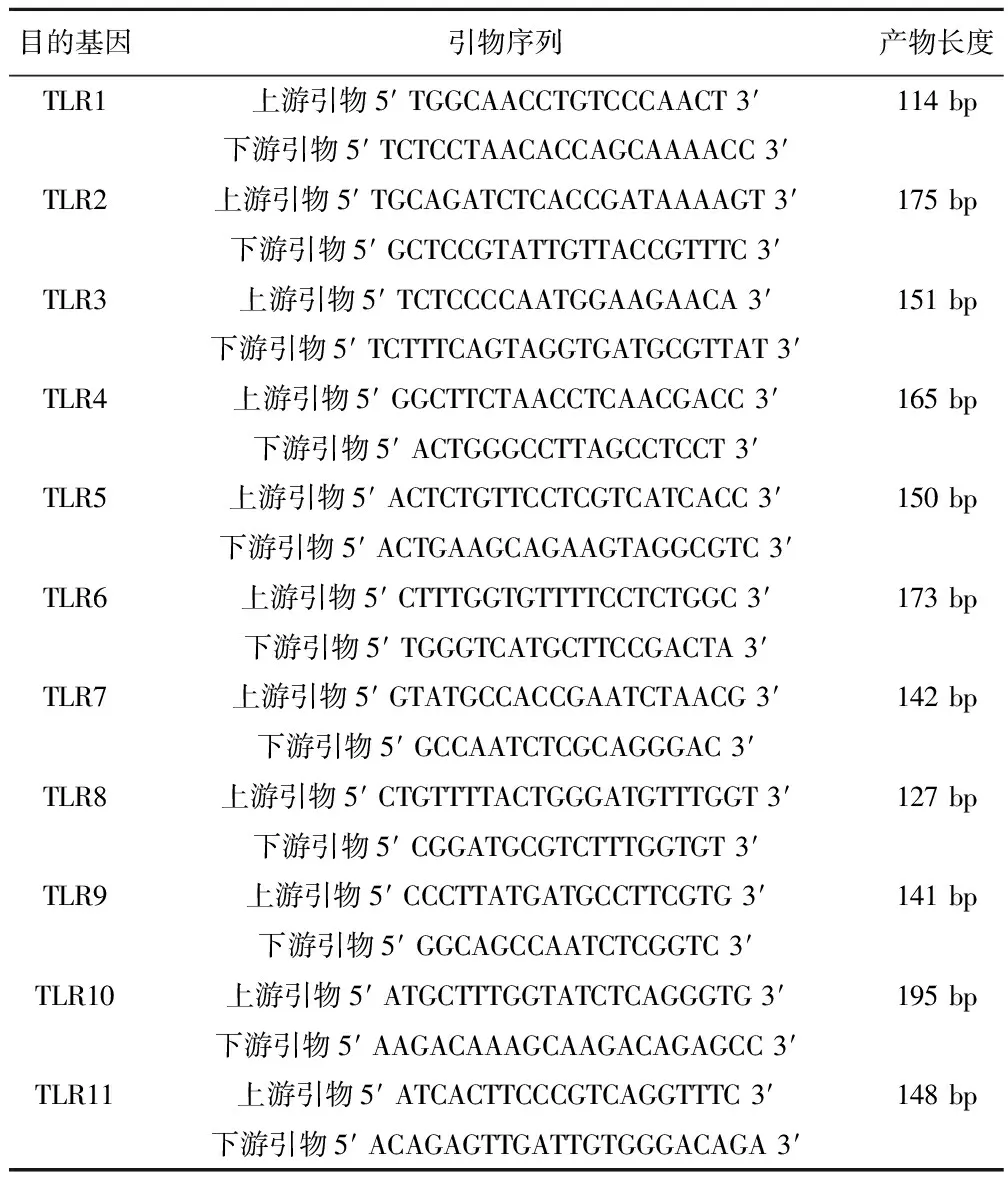
表1 PCR引物序列
1.5AS纯度鉴定 采用胶质纤维酸性蛋白(GFAP)和Hoechst的免疫荧光双标法,遵照实验操作规程,对培养的AS进行纯度鉴定,并用荧光显微镜观察、摄片。
1.6实时PCR法测定AS的TLRs表达谱 按照Trizol试剂说明书,提取AS总RNA。并用Eppendorf核酸蛋白分析仪对其定量。选择OD260/OD280在1.8~2.0的样本进行实时PCR反应。根据试剂盒说明,在0.2 ml EP管中,取总RNA0.1~0.5 μg组成反应体系,于PCR仪进行扩增。RT参数:42℃,60 min 70℃,5 min 4℃保存。PCR参数:94℃,5 min(94℃,30 s 54℃,45 s 72℃,45 s)35个循环72℃,7 min 4℃保存。用1.5%琼脂糖凝胶电泳鉴定PCR产物,电泳结束用凝胶成像系统拍摄并对结果进行分析。为了减少误差,以TLRs与β-actin条带密度之比表示TLRs的相对含量。
1.7免疫荧光双标法测定TLR4在AS中的表达定位 依据说明书,对纯化的AS进行TLR4和Hoechst免疫荧光染色,IX51倒置荧光相差显微镜下观察定位。

2 结 果
2.1AS的形状和状态 原代培养24 h后开始贴壁生长,换液后继续培养,细胞开始有细小的突起长出。随着培养时间的延长,突出越来越多、越来越长,AS所占的比例越来越大。至9~10 d细胞聚集生长,胞体呈圆形或椭圆形,有较好的折光性,突起为两极或三极,数量多,增殖旺盛。于倒置相差显微镜下观察见大量AS铺满瓶底,并融合成单层。传代后的AS生长更为活跃,一般7 d左右即可铺满瓶底,镜下观察细胞形态不规则,扁平状相互融合成片,胞质丰富(图1)。经鉴定,AS纯度>95%(图2),满足实验要求。
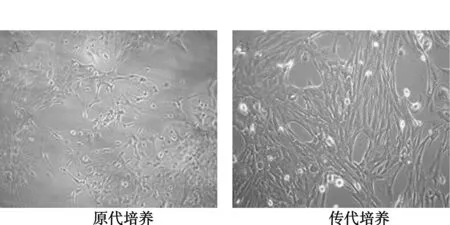
图1 大鼠皮层星形胶质细胞的形态(×200)
2.2AS TLRs表达谱 TLR1~11均有不同程度的表达,但不同的TLRs表达量不同,其中TLR4表达水平相对较高,与TLR1和TLR6相比,有统计学意义(P<0.05)。进一步实验证明,TLR4表达定位于星形胶质细胞的胞膜和胞浆,是一种跨膜受体(图3C、D、E)。因此,AS可能通过TLR4介导先天性或获得性免疫反应参与AD的炎症发病机制假说。
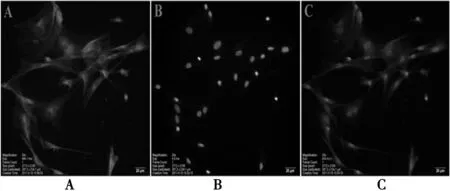
A:GFAP,AS胞质染成红色; B:Hoechst33342,所有细胞核染成蓝色; C:A和B的合并图

A:TLR1~11的琼脂糖凝胶电泳图,1~11:TLR1~11,12:β-actin,M:marker;B:TLR1~11相对定量柱形图,TLR4与TLR1和 TLR6相比:1)P<0.05; C:TLR4,AS胞膜和胞质染成绿色; D:Hoechst33342,所有细胞核染成蓝色; E:C与D合并图
3 讨 论
AD的病理特征主要表现为老年斑(SP)、神经原纤维缠结(NFTs)和神经元丢失。AD的病因和发病机制十分复杂,迄今为止,尚未完全清楚。目前普遍认为,它是各种因素累积效应的结果〔9〕,例如年龄、基因突变和免疫反应等。因此,关于AD的发病机制也存在着多种学说,如Aβ学说、Tau蛋白过度磷酸化学说、炎症和免疫学说等〔10〕。越来越多的证据表明,脑内存在的慢性炎症反应与AD的发生发展有关,在AD的发病机制中发挥重要的作用〔11~13〕。因此,提出了AD发病机制的炎症假说,包括炎症细胞、炎症因子和自由基等。而且,近来的研究〔14,15〕发现,用非甾体抗炎药(NSAIDs)美洛昔康治疗AD大鼠,可以减少脑组织内炎症因子的水平及抑制环氧合酶-2(COX-2)的表达,从而延缓和抑制AD发病过程中的炎症反应,降低AD的风险和延缓疾病的进展。
AS是中枢神经系统内分布最广泛的胶质细胞,在脑内数量最多,是神经元数量的10倍左右〔16〕。近年来,越来越多的人们关注AS,其在神经退行性疾病发病机制中作用已成为研究的热点。传统观点认为,AS为神经元提供结构及营养支持,是被动的次要角色。近年来研究表明,AS激活可合成和分泌许多炎症因子和细胞因子,参与神经系统疾病的炎症免疫反应〔17〕,在包括AD、帕金森病在内的神经退行性疾病发生和发展过程中起重要的作用〔18~22〕。
本研究将原代培养的AS经过多次传代以后,一些神经元、血管内皮细胞、小胶质细胞及成纤维细胞等逐渐减少,最终AS占主要地位。本实验中经传代后的AS生长活跃,一般7 d左右即可铺满瓶底,镜下观察细胞形态不规则,扁平状,相互融合成片,胞质丰富。GFAP是中枢神经系统中AS所独有的细胞骨架蛋白,是公认的AS特征性标记物〔23~25〕。因此,本研究中采用GFAP鉴定培养的AS纯度,结果表明,培养细胞经过3次传代以后,AS纯度达95%以上,满足实验的要求。
近年来发现的TLRs是一组新的模式识别受体(PRRs),在很多病原微生物引起的感染性疾病和炎症性损伤中的作用逐渐引起了人们的关注。TLRs表达于许多免疫和非免疫细胞,构成了先天性免疫的第一道防线。目前,有大量与胶质细胞相关的研究,对小胶质细胞的作用研究比较明确,小胶质细胞相当于中枢神经系统的单核巨噬细胞,表达TLR1~9〔26〕,由小胶质细胞激活介导的炎症反应加剧了脑内神经元的死亡,而对AS激活的作用则不够明确。
本实验采用实时PCR方法和免疫荧光技术对AS TLRs的表达进行了研究,结果表明,正常情况下,AS不同程度地表达TLR1~11,其中TLR4表达水平相对较高,而且TLR4定位于AS的胞膜和胞浆,是一跨膜受体。为后续探讨AS参与AD神经炎症的可能分子机制提供了实验基础。
4 参考文献
1Kamalvand G,Ali-Khan Z.Immunolocalization of lipid peroxidation/advanced glycation end products in amyloid A amyloidosis〔J〕.Free Radic Bio Med,2004;36(5):657-64.
2Srikanth V,Maczurek A,Phan T,etal.Advanced glycation endproducts and their receptor RAGE in Alzheimer′s disease〔J〕. Neurobiol Aging,2011;32(5):763-77.
3Hickman SE,Allison EK,El Khoury J.Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer′s disease mice〔J〕. J Neurosci,2008;28(33):8354-60.
4Miller MC,Tavares R,Johanson CE,etal.Hippocampal RAGE immunoreactivity in early and advanced Alzheimer′s disease〔J〕.Brain Res,2008;1230:273-80.
5Ghidoni R,Benussi L,Glionna M,etal.Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment〔J〕.J Neural Transm,2008;115(7):1047-50.
6Takeda K,Akira S. Toll-like receptors in innate immunity〔J〕. Int Immunol,2005;17(1):1-14.
7Chen K,Huang J,Gong W,etal.Toll-like receptors in inflammation,infection and cancer〔J〕.Int Immunopharmacol,2007;7(10):1271-85.
8Kawai T,Akira S.Signaling to NF-〔kappa〕 B by Toll-like receptors〔J〕. Trends Mol Med, 2007;13(11):460-9.
9Wan Y,Wang G,Chen SD.Genetic predisposition to inflammation:a new risk factor of Alzheimer′s disease〔J〕.Neurosci Bull,2008;24(5):314-22.
10Braak H,Del Tredici K.Alzheimer′disease:pathogenesis and prevention〔J〕.Alzheimers Dementi,2012;8(3):227-33.
11Griffin WST. Inflammation and neurodegenerative diseases〔J〕. Am J Clin Nutr,2006;83(2):470-4.
12Hoozemans JJM,Veerhuis R,Rozemuller JM,etal.Neuroinflammation and regeneration in the early stages of Alzheimer′s disease pathology〔J〕. Inte J Dev Neurosci,2006;24(2-3):157-65.
13Salminen A,Ojala J,Kauppinen A,etal.Inflammation in Alzheimer′s disease:amyloid-〔beta〕 oligomers trigger innate immunity defence via pattern recognition receptors〔J〕.Prog Neurobiol,2009;87(3):181-94.
14茅家慧,周爱玲,胡亚娥,等.美洛昔康对Aβ诱导 Alzheimer′s disease 大鼠脑内炎症损伤的影响〔J〕.中国应用生理学杂志,2010;26(1):66-70.
15in′t Veld BA,Ruitenberg A,Hofman A,etal.Nonsteroidal antiinflammatory drugs and the risk of Alzheimer′s disease〔J〕.New Engl J Med,2001;345(21):1515-21.
16Agius E,Decker Y,Soukkarieh C,etal.Role of BMPs in controlling the spatial and temporal origin of GFAP astrocytes in the embryonic spinal cord〔J〕.Dev Biol,2010;344(2):611-20.
17Benarroch EE.Neuron-astrocyte interactions:partnership for normal function and disease in the central nervous system〔J〕.Mayo clin Proc,2005;30(10):1326-38.
18Mirza B,Hadberg H,Thomsen P,etal.The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson′s disease〔J〕.Neuroscience,1999;95(2):425-32.
19Reyes JF,Reynolds MR,Horowitz PM,etal.A possible link between astrocyte activation and tau nitration in Alzheimer′s disease〔J〕. Neurobiol Dis,2008;31(2):198-208.
20Tuppo EE,Arias HR.The role of inflammation in Alzheimer′s disease〔J〕. Int J Biochem Cell B,2005;37(2):289-305.
21Wyss-Coray T.Inflammation in Alzheimer disease:driving force,bystander or beneficial response〔J〕.Nat Med,2006;12(9):1005-15.
22Aloisi F,Ria F,Adorini L.Regulation of T-cell responses by CNS antigen-presenting cells:different roles for microglia and astrocytes〔J〕. Immunol Today,2000;21(3):141-7.
23Eng LF,Ghirnikar RS,Lee YL.Glial fibrillary acidic protein:GFAP-thirty-one years (1969-2000)〔J〕.Neurochem Res,2000;25(9):1439-51.
24Gomes FCA,Paulin D,Moura Neto V.Glial fibrillary acidic protein (GFAP):modulation by growth factors and its implication in astrocyte differentiation〔J〕.Brazl J Med Biol Res,1999;32(5):619-31.
25Miljkovi D,Timotijevi G,Stojkovi MM.Astrocytes in the tempest of multiple sclerosis〔J〕.FEBS Lett,2011;585(23):3781-8.
26Carpentier PA,Duncan DAS,Miller SD.Glial toll-like receptor signaling in central nervous system infection and autoimmunity〔J〕.Brain Behav Immu,2008;22(2):140-7.
