新型含吲哚酮环的螺吡喃化合物的合成*
2014-08-29魏文斌赵祖建
魏文斌,赵祖建
(1.南京理工大学 泰州科技学院,江苏 泰州 225300;2.太仓中化环保化工有限公司,江苏 太仓 215400)
·快递论文·
新型含吲哚酮环的螺吡喃化合物的合成*
魏文斌1,赵祖建2
(1.南京理工大学 泰州科技学院,江苏 泰州 225300;2.太仓中化环保化工有限公司,江苏 太仓 215400)
以对取代苯胺为原料,将吡喃环引入吲哚-2-酮结构中,设计并合成了一系列新型的含吲哚酮环的螺环化合物——5′-乙酰基-2′-氨基-1,2-二氢-6′-甲基-5-取代基-2-氧代螺[3H-吲哚-3,4′-(4H)吡喃]-3′-甲腈,其结构经1H NMR和元素分析表征。
吲哚酮;吡喃;螺环化合物;合成
含有吲哚酮母核的化合物具有抗肿瘤[1]、消炎镇痛以及作为受体络氨酸激酶抑制剂等多种生物活性,在新药设计中常被作为有效的活性基团。3-取代吲哚-2-酮是近几年研究和开发的一类抗肿瘤药物,具有高效、选择性好的特点,是一种极有发展前景的抗肿瘤药物。闵真立等[2]合成了一系列3-取代苯基亚甲基吲哚-2-酮衍生物,研究发现部分衍生物具有抗肿瘤活性。酮基布洛芬的重要中间体就含有3-甲基吲哚-2-酮和5-苯甲酰基-3-甲基吲哚-2-酮两个化合物[3]。
含吡喃环的杂环化合物广泛分布于植物和生物碱中,具有良好的生物活性,成为医药合成的重要母体。Mina Saeedi[4]等以丙二腈、萘并乙二酮(苊醌)和乙酰丙酮为原料,在三乙胺催化下用乙醇做溶剂回流反应合成了一种具有抗高血压功效的螺吡喃化合物。[吲哚-吡喃]螺环化合物被广泛地用在肌肉弛缓药、催眠药和消炎药的合成中。3-二氰基亚甲基吲哚-2-酮和吡咯啉化合物进行Micheal加成是合成[吲哚-吡喃]螺环化合物[5]的方法之一。
本文以4-取代苯胺(1a~1d)为起始原料,在硫酸钠饱和溶液中与水合氯醛和盐酸羟胺通过Sandmeyer反应制得N-(4-取代苯基)-2-肟基乙酰胺(2a~2d);2在浓硫酸作用下关环,水解得5-取代靛红(3a~3d);3与活泼亚甲基化合物丙二腈通过克诺文格尔反应得2-(5-取代二氢吲哚-2-酮-3-亚基)丙二腈(4a~4d);4与乙酰丙酮通过迈克尔加成反应合成了一系列新型的含吲哚酮环的螺环化合物5′-乙酰基-2′-氨基-1,2-二氢-6′-甲基-5-取代基-2-氧代螺[3H-吲哚-3,4′-(4H)吡喃]-3′-甲腈(5a~5d,Scheme 1),其结构经1H NMR和元素分析表征。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪(温度未校正);BRUKER DRX-300型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Flash EA1112型元素分析仪。
1a~1d和盐酸羟胺,化学纯;其余所用试剂均为分析纯。
1.2 合成
(1)2a~2d的合成(以2a为例)
在烧杯中依次加入去离子水100mL,浓盐酸11mL(0.11mol),苯胺(1a)10.23g(0.11mol),搅拌至透明得溶液A。
在四口烧瓶中加入水300mL和硫酸钠80g~120g,于40℃搅拌使其溶解;加入水合氯醛20g(0.12mol),搅拌致其溶解;滴加溶液A(边滴加边有大量白色絮状沉淀生成),滴毕,于70℃反应30min。迅速加入盐酸羟胺23g(0.33mol),搅拌30min,于85℃反应至终点(TLC检测),冷却至室温,过滤,滤饼干燥得淡黄色固体N-苯基-2-肟基乙酰胺(2a)。
分别用1b~1d替代1a,用类似方法合成2b~2d。
(2)3a~3d的合成(以3a为例)
在四口烧瓶中加入浓硫酸50mL,加热至60℃时缓慢加入2a16.4g (0.10mol),于85℃~90℃反应30min,冷却至室温,倒入500mL冰水中,搅拌过滤,滤饼用水洗涤至中性后用无水乙醇重结晶得橘红色粉末3a12.05g。
分别用2b~2d替代2a,用类似方法合成橘红色粉末3b~3d。
3a:收率82.0%,m.p.203℃(202℃[6])。
3b:收率81.3%,m.p.186℃~187℃(186℃~187℃[7]);1H NMRδ:2.28(s,3H,CH3),6.80~7.42(m,3H,ArH),10.94(s,1H,NH)。
3c:收率84.7%,m.p.220℃~222℃(221℃~222℃[8]);1H NMRδ:6.89~7.48(m,3H,ArH),11.02(s,1H,NH)。
3d:收率88.1%,m.p.247℃~248℃(246℃~247℃[7]);1H NMRδ:6.64~7.61(m,3H,ArH),11.12(s,1H,NH)。
(3)4a~4d的合成(以4a为例)
在四口烧瓶中加入3a1.47g(10.0mmol),丙二腈0.66g(0.01mol)和无水乙醇20mL,搅拌下滴加5滴吡咯烷,回流反应至终点(TLC检测)。冷却至室温,搅拌过滤,滤饼用乙醇重结晶,干燥得黑色粉末4a1.53g。
分别用3b~3d替代3a,用类似方法合成黑色粉末4b~4d。
4a:收率78.5%,m.p.235℃~236℃(236℃~238℃[9]);1H NMRδ:6.90~7.88(m,4H,ArH),11.18(s,1H,NH)。
4b:收率80.6%,m.p.256℃~260℃(260℃[10]);1H NMRδ:2.40(s,3H,CH3),6.83~7.67(m,3H,ArH),11.08(s,1H,NH)。
4c:收率79.9%,m.p.246℃(230℃[11]);1H NMRδ:6.93~7.73(m,3H,ArH),11.34(s,1H,NH)。
4d:收率81.4%,m.p.208℃~210℃;1H NMRδ:6.96~7.78(m,3H,ArH),11.34(s,1H,NH)。
(4)5a~5d的合成(以5a为例)
在四口烧瓶中加入4a1.95g(10.0mmol)和乙醇50mL,搅拌下依次加入乙酰丙酮1.02mL(10.0mmol)和三乙醇胺1.32mL(10.0mmol),回流反应至终点(TLC检测)。冷却至室温,抽滤,滤饼依次用正己烷、乙醇洗涤,干燥得乳白色粉末5a1.93g。
分别用4b~4d替代4a,用类似方法合成乳白色粉末5b~5d。
5a:收率65.4%,m.p.239℃~240℃(240℃[12]);1H NMRδ:2.08(s,3H,CH3),2.28(s,3H,OCH3),6.76~6.79(d,J=9.0Hz,1H,ArH),6.89~6.94(t,1H,ArH),7.02~7.04(d,J=6.0Hz,1H,ArH),7.08(s,2H,NH2),7.13~7.19(t,1H,ArH),10.18(s,1H,NH);Anal.calcd for C16H13N3O3:C 65.46,H 4.75,N 14.14;found C 65.08,H 4.44,N 14.23。
5b:收率62.2%,m.p.256℃~260℃;1H NMRδ:2.08(s,3H,CH3),2.28(s,3H,OCH3),2.50(s,3H,ArCH3),6.65~6.97(m,3H,ArH),7.06(s,2H,NH2),10.27(s,1H,NH);Anal.calcd for C17H15N3O3:C 65.88,H 5.02,N 13.55;found C 66.01,H 4.89,N 13.58。
5c:收率68.3%,m.p.255℃~257℃;1H NMRδ:2.08(s,3H,CH3),2.28(s,3H,OCH3),6.74~7.01(m,3H,ArH),7.16(s,2H,NH2),10.37(s,1H,NH);Anal.calcd for C16H12N3O3F:C 61.49,H 3.80,N 13.77;found C 61.34,H 3.86,N 13.41。
5d:收率66.9%,m.p.208℃~210℃;1H NMRδ:2.19(s,3H,CH3),2.35(s,3H,COCH3),6.77~7.11(m,2H,ArH),7.17~7.21(d,3H,ArH,NH2),10.18(s,1H,NH);Anal.calcd for C16H12N3O3Cl:C 58.30,H 3.72,N 12.71;found C 58.28,H 3.67,N 12.74。
2 结果与讨论
2.1 脱水关环温度对3a~3d收率的影响
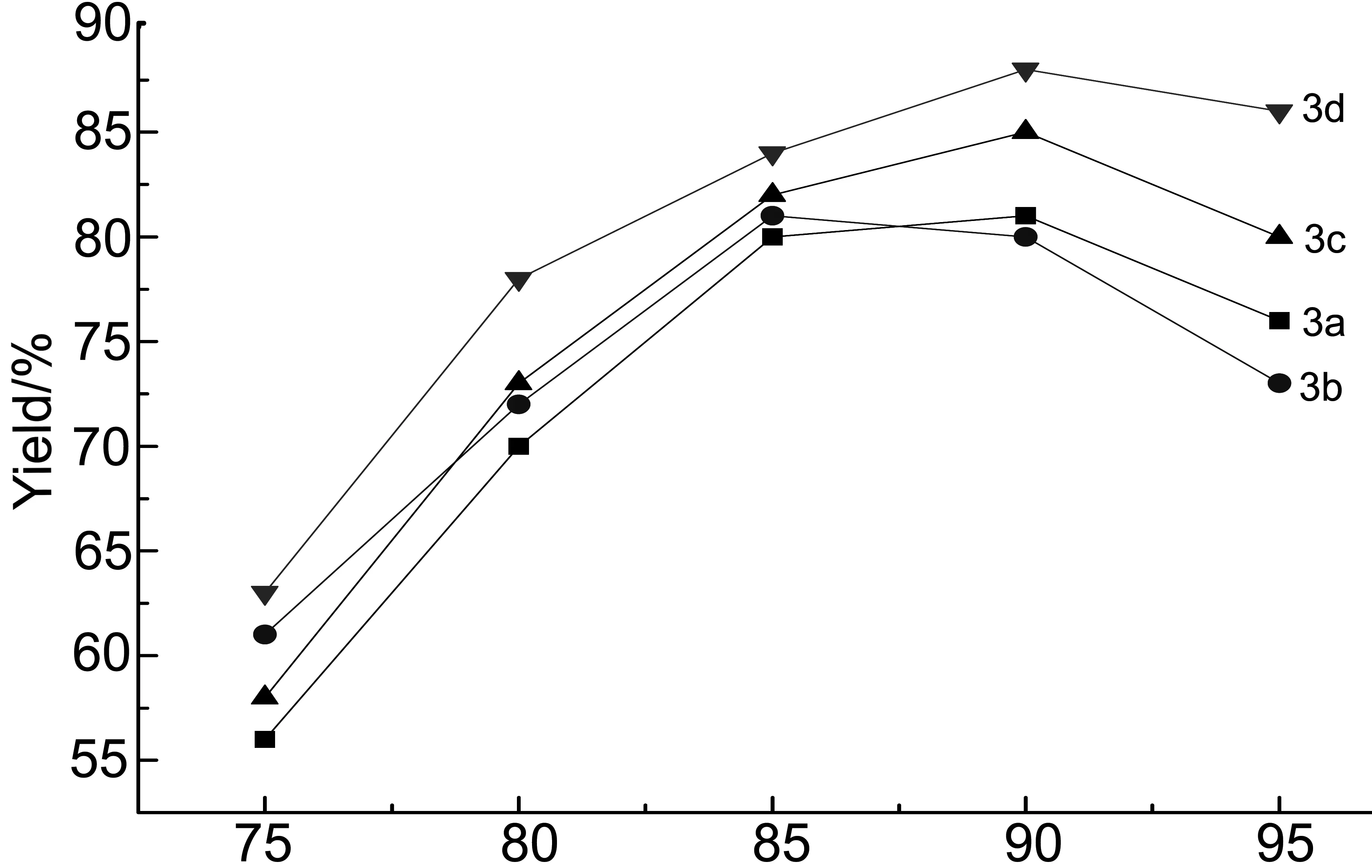
脱水关环温度对3a~3d收率的影响见图1。由图1可见,浓硫酸关环脱水温度对3a~3d的收率影响较大。当反应温度75℃时,此时温度较低原料反应不剧烈,脱水关环程度不够,收率较低;当升高温度到90℃时,收率较高(均达到80%以上);继续升高反应温度,产物收率开始下降。这是因为温度太高,副产物增多所致。因此,最佳脱水关环温度为85℃~90℃。
Temperature/℃
2.2 构效分析
5a~5d的收率大小顺序为5c>5d>5a>5b,可能的原因是反应物4a~4d的5-位取代基对其3-位C原子活性的影响。当取代基为强吸电子基时,有利于碳负离子进攻3-位C促进Micheal加成反应,取代基吸电子能力大小顺序F>Cl>H>CH3,所以证实了收率大小顺序与上述结果一致。
3 结论
本文以对取代苯胺为原料,在不改变吲哚-2-酮结构的情况下,将吡喃环引入到吲哚-2-酮的3-位上,制备了一系列新型的螺环化合物。
[1] Ullrich A,Shlessinger J.Signal transduction genetics of cancer by receptors with tyrosine kinase activity[J].Cell,1990,61(2):203-212.
[2] 闵真立,姜凤超,张奇.3-取代吲哚酮类化合物的合成及抗肿瘤活性[J].中国药物化学杂志,2005,(13):129-32.
[3] 户业丽,管春生,苏健宇.重氮还原法合成酮基布洛芬[J].湖北化工,2002,17(2):23-24.
[4] Mina Saeedi,Majid M Heravi,YahyaS Beheshtiha,etal.One-pot three-component synthesis of the spiroacenaphthylene derivatives[J].Tetrahedron,2010,66(29):5345-5348.
[5] Dandia,Taneja,Gupta,etal.An efficient procedure for the synthesis of spiro {3H-indole-3,4′-(1′H)-pyrano [2,3-c]pyrrole}-5′-carbonitriles using solid inorganic supports and microwave activation[J].Synthetic Communications,1999,29(13):2323-2335.
[6] Taylor E C,Eckroth D R.Mechanism of conversion ofo-nitrobenzoyldiazomethane intoN-hydroxyisatin[J].Tetrahedron,1964,20(9):2059-2064.
[7] Da Silva J F M,Garden S J,Pinto A C.The chemistry of isatins:A review from 1975to 1999[J].Journal of Brazilian Chemical Society,2001,12(3):273-324.
[8] Lackey K,Besterman J M,Fletcher W,etal.Rigid analogs of camptothecin as DNA topoisomerase inhibitors[J].J Med Chem,1995,38(6):906- 911.
[9] Adam,Jean Marie.Nucleophilic reactions of 2-phenylindolenin-3-ones[J].Helvetica Chimica Acta,1984,67(8):2186-2191.
[10] Anderson D M W.Some condensation products of malononitrile[J].Journal of the Chemical Society,1961,4705-4711.
[11] Joshi,Krishna C.Studies in spiro-heterocycles.Part-XII.Synthesis of some fluorine containing spiro{3H-indole-3,4′(4H)-pyrano[2,3-d]pyrimidine}-2,5′,7′(1H)-triones as CNS agents[J].Journal of the Indian Chemical Society,1988,65(3):202-204.
[12] L A Shemchuk,V P Chernykh,R G Red'kin.Synthesis of fused 2′-amino-3′-R-spiro-indole-3,4′-pyran-2(1H)-ones[J].Russian Journal of Organic Chemistry,2008,44(12):1789-1794.
SynthesisofNovelSpiropyranCompoundswithOxindoleRing
WEI Wen-bin1,ZHAO Zu-jian2
[1.Taizhou Institute of Technology,Nanjing University of Science and Technology,Taizhou 225300,China;2.Sinocheu Environmental Protection Chemicals(Taicang)Co.,LTD,Taicang 215400,China]
Pyran was introduced into 3-position of oxindole,then a series of novel spiropyran compounds with oxindole ring were designed and synthesized using substituted-aniline as the starting materials.The structures were characterized by1H NMR and elemental analysis.
oxindole;pyran;spiro compound;synthesis
2013-03-28;
2014-06-06
魏文斌(1984-),男,汉族,宁夏隆德人,助教,硕士研究生,主要从事药物合成的研究。Tel.0523-86150081,E-mail:wenbinwei@yeah.net
O626.31
A
1005-1511(2014)04-0513-03
