含氟双二苯乙炔液晶的合成及液晶性
2014-08-21范程士罗忠林刘克刚闻建勋
范程士 罗忠林 刘克刚 闻建勋
(1.上海天问化学有限公司,上海200232;2.University Hospital Basel,Intensive Care Unit,Petersgraben,Basel44031)
为了满足液晶平板显示发展的各项要求,如响应速度快及器件视角宽的液晶材料,开发了具有不同特点显示模式的显示器。毫无疑问,具有正或者负的介电各向异性的向列型液晶材料是液晶显示材料家族十分重要的成员。正性向列型液晶可用于标准的TFT-LCD(薄膜晶体管-液晶)显示及IPS-LCD(面内开关-液晶)显示(电场2极在同一块板面上,施加电场使极性分子在电场中改变方向)。液晶分子轴方向与垂直分子轴方向的介电常数之差Δε为负数的称为负性液晶,既可以在VA-TFT LCD(分子垂直排列的薄膜晶体管液晶显示)模式中也可以在IPS模式中得到广泛应用。
如果含氟向列型液晶具有高的双折射性能,它们还可以在许多其他显示技术方面得到重要应用。例如,胆甾相液晶具有电场为0时的多稳定相态织构现象,反射式胆甾液晶显示,由于不需要偏振片及背光源,因此具有高反射能力及宽视角,特别适用于电子书籍及商业广告等领域。还有在高技术的光子学应用领域,例如制造快速的快门、宽带滤波器及全息摄影器件等。尤其是光子学领域的器件需要黏度低及双折射大的混合液晶。这就是本工作的目的。
一般来说,没有取代基的双二苯乙炔有非常高的双折射,但缺点是熔点高、对紫外线敏感、化学稳定性差。为此,本工作在结构上进行改造,在液晶核骨架上引入全氟亚苯基基团,对降低熔点、降低黏度有利,提高了化学稳定性,氟原子的范德华半径与原子差别不大,保持非常宽的液晶温度区域。侧方向的2,3-二氟取代的导入,引入了2个强吸引电子的基团,结果形成优秀的负性向列型含氟液晶。可以分别以VA、IPS 2种不同工作模式应用于许多需要高速相应的光学器件中。优良的液晶原材料应具备高清亮点、液晶相范围宽、高双折射、黏度低、脂溶性好等特征,其中分子线性共轭性强的液晶化合物具有高双折射率,例如多芳环类、炔类和多炔类化合物。
在上述化合物中,双二苯乙炔类液晶的双折射率n都在0.4以上,但是由于分子的共轭性很强造成此类化合物熔点很高,因此人们往往通过引入侧基来降低熔点和黏度。2000年,C SHsu等人报道利用Sonogashira反应合成了含烷基边链系列化合物,其分子特点是2端为烷基链,侧向为短碳链烷基[1]。
氟原子的电负性强、体积小及脂溶性好等特点,在医药和材料上具有很广泛的应用。笔者等在双炔键的基础上引入氟原子及四氟苯环特征基元以期降低熔点,增加向列相温度范围等特点设计、合成了一系列新颖含氟双二苯乙炔类液晶,并对其相变性质进行研究。
结构通式:
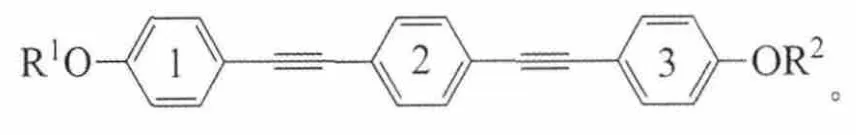
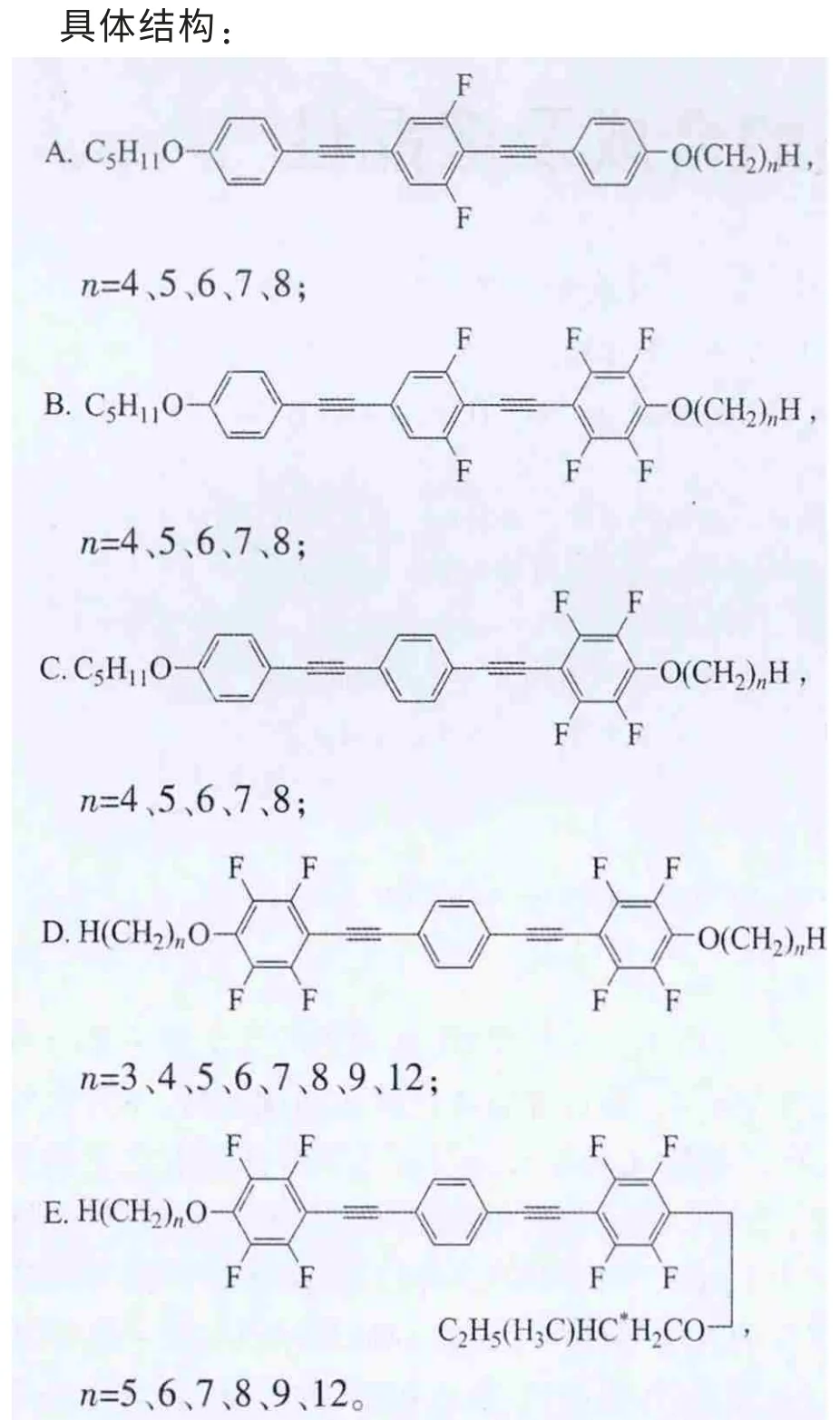
上述5类化合物的共同特点是:含有3个苯环,每2个相邻的苯环之间以炔键连接,2端苯环末端以烷氧基链结束。其中A、B、C系列化合物的特点是苯环1的烷基R1链固定为正戊烷C5H11,另一端烷基R2链随着碳原子数的变化具有不同长度[2-3]。它们的不同之处在于侧边苯环不同位置上引入氟原子:A系列是中间苯环2的2侧对称位置2个氢原子被2个氟原子取代;B系列是在A系列基础上,靠近变化末端烷基的苯环3氢原子全部被氟原子取代;C系列是只有苯环3全氟代;D系列拥有对称的结构,苯环1、3全氟代,2端具有相同的烷基链[4]。相对于D系列,E系列一端烷基链固定为手性戊基,而另一端为变化的正烷氧基链[5]。
1 化合物的合成路线
1.1 A、B、C系列
A、B、C系列化合物的合成路线:

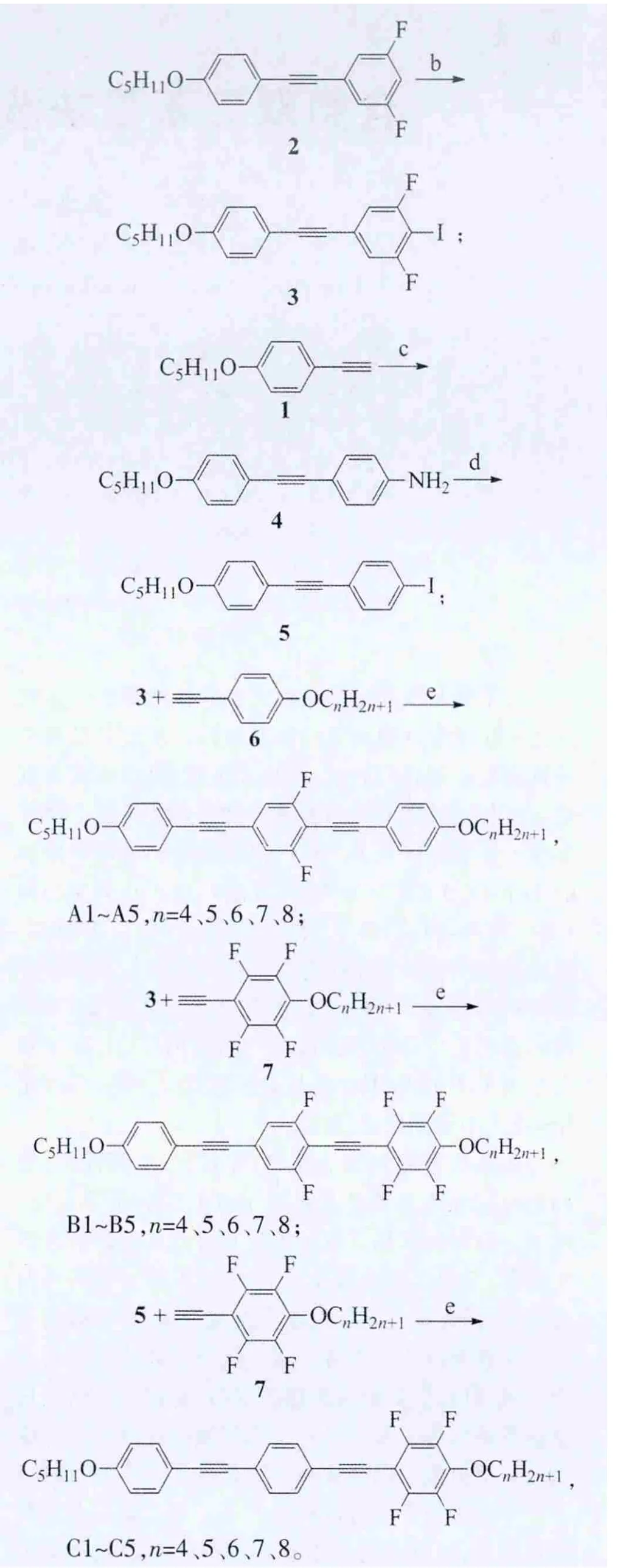
合成此类化合物的关键步骤是在II价钯和碘化亚铜催化下,炔键化合物和碘化物的偶联反应。
化合物3的制备:以正戊氧基苯乙炔1为起始原料,在Pd(PPh3)2Cl2和CuI的催化下和3,5-二氟-1-碘苯偶联得到中间体2,化合物2在正丁基锂的作用下和碘反应得到关键中间体3。
化合物5的制备:同样以正戊氧基苯乙炔1为起始原料,在Pd(PPh3)2Cl2和CuI的催化下和对碘苯胺偶联得到中间体4,此化合物在亚硝酸钠、浓盐酸的氧化下得到偶氮化合物中间体,此中间体进一步和碘化钾反应生成化合物5。
在Pd(PPh3)2Cl2和CuI的催化下,4-正烷氧基苯乙炔6和碘化物3偶联得到目标化合物A;4-正烷氧基-2,3,5,6-四氟化合物7和碘化物3偶联得到目标化合物B;4,-烷氧基-,3,5,6-四氟化合物7和碘化物5偶联得到目标化合物C。
反应试剂和条件:a)3,5-二氟-1-碘苯,Pd(PPh3)2Cl2,CuI,Et3N;b)n-BuLi,I2,-78℃~室 温;c)4-碘 苯胺,Pd(PPh3)2Cl2,CuI,Et3N;d)NaNO2,浓HCl,KI,四氢呋喃;e)Pd(PPh3)Cl2,CuI,Et3N。
1.2 D系列
D系列化合物的合成路线:
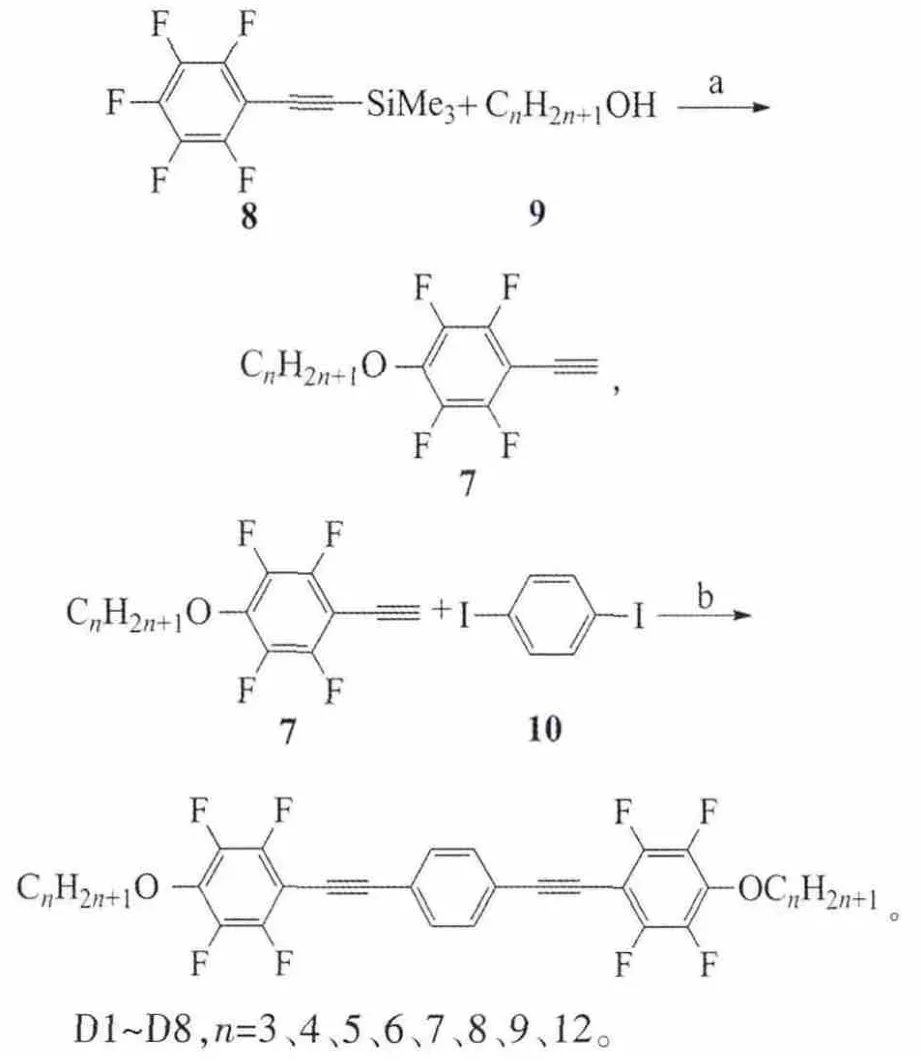
三甲基硅基五氟苯乙炔8在碳酸钾的作用下和不同链长的正烷基伯醇发生反应得到中间体7,此炔基化合物在Pd(PPh3)2Cl2和CuI(碘化亚铜)的催化下和1,4-二碘苯发生双偶联反应得到目标化合物D。
反应试剂和条件:a)K2CO3,N,N-二甲基甲酰胺(DMF),室温;b)Pd(PPh3)2Cl2,CuI,Et3N。
1.3 E系列
和上述提到的目标化合物的制备基本相似,E系列化合物的合成路线:

化合物8在碳酸钾的作用下和手性醇11反应得到末端炔12,此化合物在Pd(PPh3)2Cl2和CuI的催化下和对溴碘苯发生偶联反应生成溴化物13,同样的条件下此溴化物和末端炔7偶联提供目标分子E。
反应试剂和条件:a)K2CO3,DMF,室温;b)Pd(Ph3)2Cl2,CuI,Et3N。
2 化合物的相变
通过偏光显微观察和差示扫描量热法(DSC)对上述5个系列的液晶化合物的结构和性质进行系统分析。
2.1 A、B、C系列双二苯乙炔类化合物
A、B、C系列化合物的相变温度如表1所示,所有的化合物都呈现单一的向列相[2-3]。
A系列化合物拥有相同的结构主体,它们的不同之处在于一段烷氧基链的长度不同。总的来说,此类化合物显示出很高的清亮点(从191~214℃);同时它们都含有很宽的向列相相变范围(>97℃);另外,熔点和清亮点都随着末端烷氧基链碳原子数的增加而逐渐降低。
B系列化合物含有1个四氟苯基官能团,此类化合物同样显示高的清亮点(从176~201℃);同时每一个化合物含有宽的相变范围(>74℃);另外,熔点和清亮点都随着末端烷氧基链碳原子数的增加而逐渐降低。
C系列化合物中间苯环没有氟原子取代,此类化合物和A、B系列同样拥有很高的清亮点(从184~211℃);同时每一个化合物含有很宽的相变范围(>83℃);熔点和清亮点随着末端烷氧基链碳原子数的增加先逐渐降低后增加。
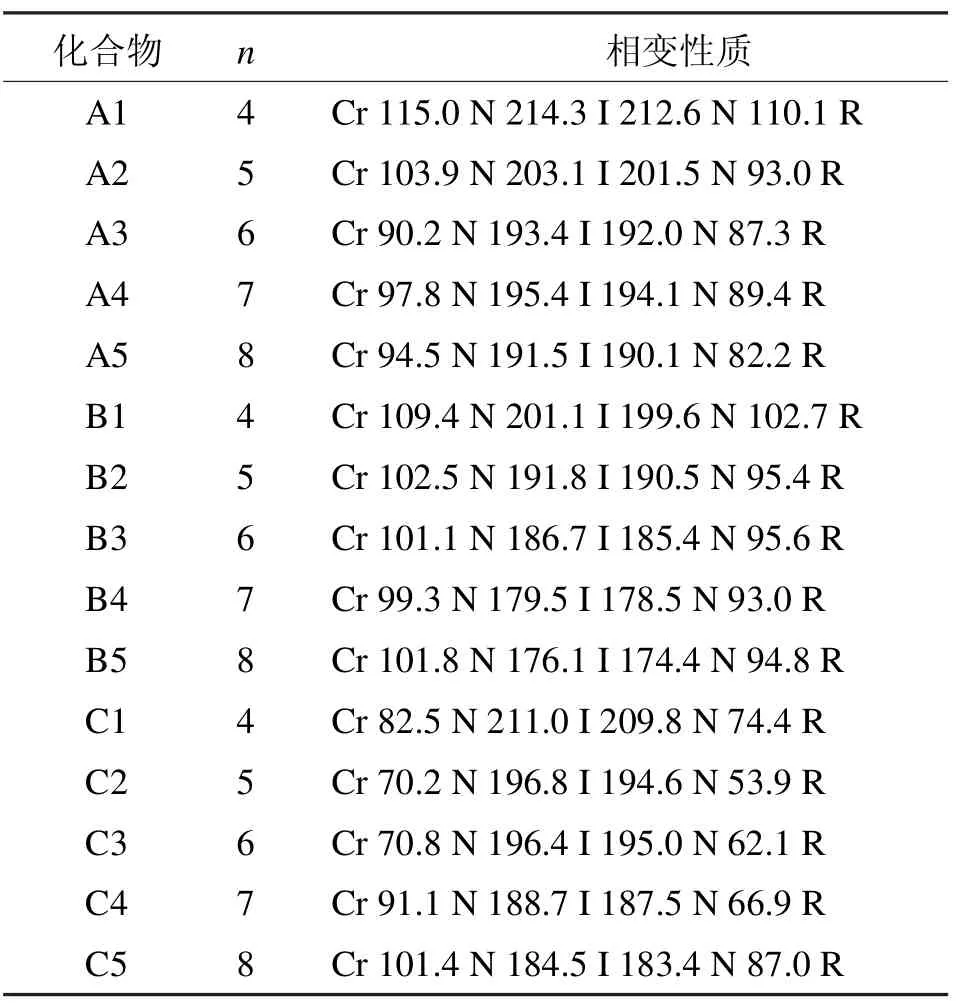
表1 A、B、C系列含氟双二苯乙炔类液晶的相变性质Tab 1 Phase transition properties of liquid crystal of A,B,C series bisdiphenylacetylene which containing fluoride
通过A、B、C 3个系列化合物的相变数据分析可以看出,侧面氟原子的取代对液晶的相变性质有很大的影响。在相同的末端烷氧基链的情况下,B系列化合物的熔点最低,而C系列的最高,清亮点则呈A>C>B趋势。双二苯乙炔类液晶共轭性很强从而拥有高的热稳定性;当氟原子引入这个结构,因为氟原子的高电负性,它们之间或和氢原子之间的相互作用造成扭曲效应从而增加分子的厚度,熔点和清亮点从而降低。
2.2 D系列双二苯乙炔类液晶
D系列化合物的相变性质如表2所示[4]。
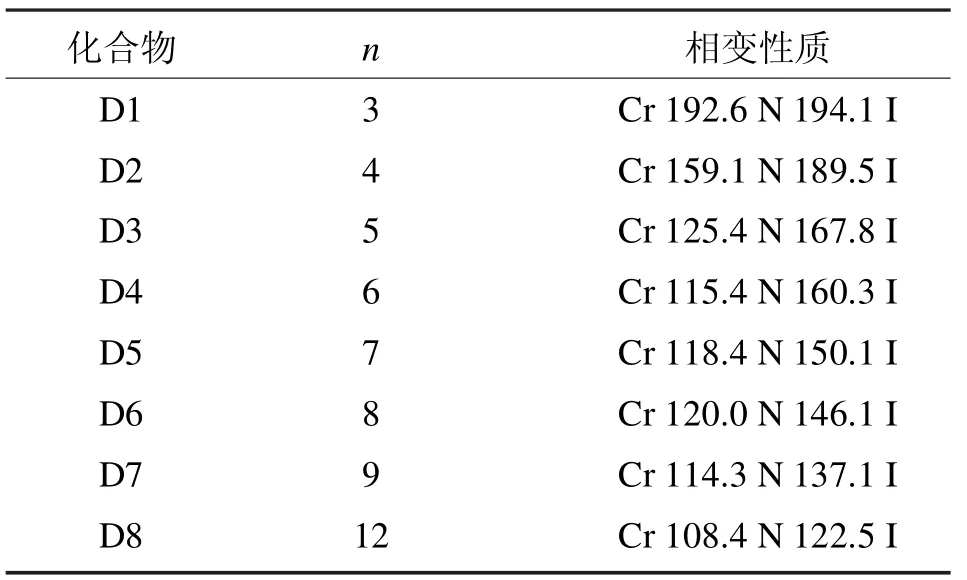
表2 D系列含氟双二苯乙炔类液晶的相变性质Tab 2 Phase transition properties of liquid crystal of D series bisdiphenylacetylene which containing fluoride
通过数据分析,8个化合物都显示单一的向列相,碳链的长度和相变温度呈现正常的奇偶效应,并且随着烷氧基链的增加,熔点和清亮点都逐渐降低,另外,向列相的范围先增加(烷氧基链碳原子个数n=3~6),随后逐渐降低(烷氧基链碳原子个数n=7~12)。和已知文献报道的苯环不含有氟原子并含有相同末端烷氧基链的双二苯乙炔类液晶相比,D系列化合物熔点和清亮点降低,而且不支持近晶相(不稳定)[5]。
2.3 E系列双二苯乙炔类液晶
E系列化合物的相变性质如表3所示[6]。
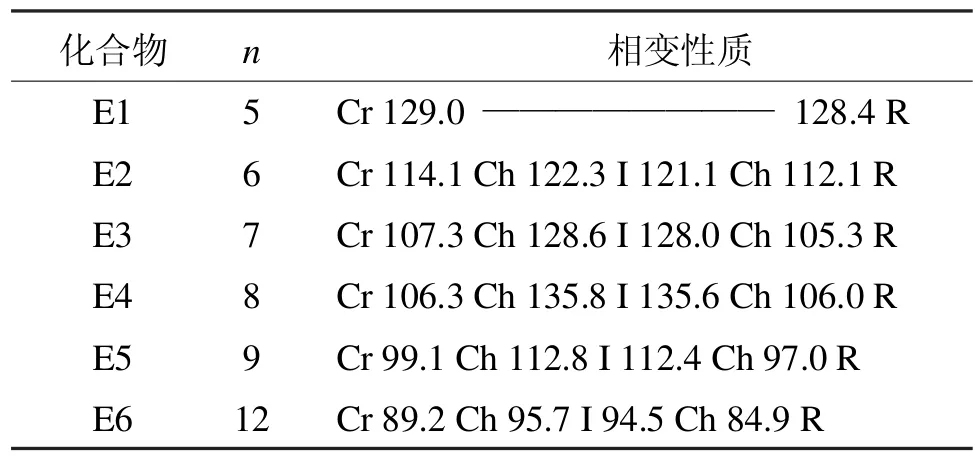
表3 E系列含氟双二苯乙炔类液晶的相变性质Tab 3 Phase transition properties of liquid crystal of E series bisdiphenylacetylene which containing fluoride
除了烷氧基链为正戊烷氧基的化合物不呈现液晶态,其他化合物均具有单一的胆甾相。就末端烷氧基链的影响考虑,首先熔点随着链长的增加而降低;而胆甾相变范围是先增加(n=6-8)后降低。此类化合物的相变性质和D系列化合物相比,相变范围变窄这说明支链化烷氧基链引起液晶的稳定性降低。
3 典型中间体和目标化合物的合成
3.1 4-正戊氧基-3′,5′-二氟二苯乙炔(2)
在一干燥的250 mL三颈瓶氮气保护下加入4-正戊氧基苯乙炔5.0 g(26.6 mmol)、3,5-二氟-1-碘苯5.32 g(22.2 mmol)、Pd(PPh3)2Cl20.2 g(0.29 mmol)和CuI 0.1 g,随后加入无水Et3N 100mL。反应混合液在30~40℃搅拌24 h。TLC跟踪反应直至完全后,沉淀过滤乙醚萃取,有机相水洗,硫酸镁干燥。真空除掉溶剂,硅胶柱层析得到白色固体7.23 g,产率91%。
1H NMR(CDCl3,TMS)(δ/ppm):7.56~7.18(m,7H),4.01(t,2H,J=5.4 Hz),1.90~1.34(m,6H),1.02(t,3H,J=3.7 Hz);19FNMR(CDCl3,TFA)(δ/ppm):32.6(m,2F)。
3.2 4-正戊氧基-3′,5′-二氟-4′-碘二苯乙炔(3)
4-正戊烷氧基-3′,5′-二氟二苯乙炔7.32 g(24.1mmol)溶解在84mL的THF中并冷却到-78℃。向上述溶液中慢慢滴加13.3mL正丁基锂(2 mol/L),此混合溶液在上述温度下继续搅拌1.5h。碘溶解在THF中,慢慢滴加到体系中,反应混合液升至室温并继续搅拌10h。随后饱和氯化铵萃灭此反应。正常后处理得到粗产品,柱层析给出白色晶体5.8g,产率57%。
1HNMR(CDCl3,TMS)(δ/ppm):7.48~6.82(m,6H),3.98(t,2H,J=5.5Hz),2.01~1.27(m,6H),0.95(t,3H,J=3.6Hz);19FNMR(CDCl3,TFA)(δ/ppm):16.5(m,2F)。
3.3 4-正戊氧基-4′-氨基二苯乙炔(4)
合成路线和化合物4-正戊氧基-3′,5′-二氟二苯乙炔相似,得到化合物5.84g,产率79%。
1HNMR(CDCl3,TMS)(δ/ppm):7.42~6.61(m,8H),3.88(t,2H,J=5.5Hz),3,61(br,2H),1.79~1.27(m,6H),0.86(t,3H,J=3.7Hz)。
3.4 4-正戊氧基-4′-碘二苯乙炔(5)
4-正戊烷氧基-4′-氨基二苯乙炔2.8g(10.04 mmol)溶解在15mL的THF中并冷却至0℃,浓盐酸5.5mL和质量分数40%的亚硝酸钠水溶液6.1 mL组成的混合液加入到上述溶液中,此体系在0℃下搅拌30min,然后加入6mol/L的KI水溶液17.3 mL,继续搅拌3h,饱和硫代硫酸钠20mL淬灭反应,正己烷萃取反应,正常的后处理,粗产品后处理得到白色晶体1.8g,产率50%。
1HNMR(CDCl3,TMS)(δ/ppm):7.42~7.72(m,8H),3.87(t,2H,J=5.4Hz),1.17~1.79(m,6H),0.86(t,3H,J=3.8Hz)。
3.5 1,3,-二氟-2-{2-[4-(正辛氧基)苯基]乙炔基}-5-{2-[4-(正戊氧基)苯基]乙炔基}苯(A5)
以化合物3和化合物6为偶联原料,反应条件和过程如下面化合物的合成路线相似,得到目标化合物。
1HNMR(CDCl3,TMS)(δ/ppm):6.80~7.57(m,4H),3.99(t,4H,J=5.9Hz),0.85~1.99(m,24H);19FNMR(CDCl3,TFA)(δ/ppm):31.0(m,2F);MS(m/z):528(M+,100.0%),元素分析(EA):计算值:C79.52,H 7.24,F7.19;实际值:C79.60,H7.22,F7.18。
ABC系列其他化合物合成路线和此化合物相似。
3.6 1,4-二[(2,3,5,6-四氟-4-正辛烷苯基)乙炔基]苯(D6)
在一干燥50mL三颈瓶,氮气保护下加入4-正辛氧基-2,3,5,6-四氟苯乙炔1.0g(3.3mmol)、对二碘苯0.55g(1.66mmol)、Pd(PPh3)2Cl230mg(0.043mmol)和CuI17mg(0.089mmol),随后加入无水Et3N15mL。反应混合液加热回流8h。TLC跟踪反应直至完全后,沉淀过滤乙醚萃取,有机相水洗,硫酸镁干燥。真空除掉溶剂,硅胶柱层析得到白色固体1.05g,产率95%。熔点120.0℃。
1HNMR(CDCl3,TMS)(δ/ppm):7.57(s,4H),4.30(t,4H,J=6.3Hz),0.74~2.00(m,30H);19FNMR(CDCl3,TFA)(δ/ppm):61.0(d,4F,J=18.6Hz),80.3(d,4F,J=18.6Hz);MS(m/z):678(M+,87.33%),454(100.00%),IR(KBr):2910、2840、1640、1520、1490、1440、1390、1140、1125、1020、1005、985、840、695、550cm-1;EA:C38H38F8O2,计算值:C67.26,H5.60,F22.42;实际值:C67.43,H5.72,F22.40。
D系列其他化合物合成路线和此化合物相似。
3.7 4-[(s)-2-甲基丁氧基]-2,3,5,6-四氟苯乙炔(12)
化合物三甲基硅基五氟苯乙炔6.0g(22.7 mmol)、碳酸钾9.0g(65.1mmol)、(s)-2-甲基丁醇4.0g(45.5mmol)溶解在12mL的DMF中,此反应体系在40℃条件下搅拌46h然后60℃继续搅拌6 h。常规后处理,粗产品柱层析得到产物为浅黄色液体5.40g(90.3%)。
1HNMR(CCl4,TMS)(δ/ppm):3.94(d,2H,J=6.0 Hz),3.34(s,1H),0.82~1.90(m,9H);19FNMR(CCl4,TFA)(δ/ppm):60.47(d,2F,J=18.8Hz),80.47(d,2F,J=18.8Hz)。
3.8 1-(4-溴苯基)-2-[4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基]乙炔(13)
在一干燥100mL三颈瓶,氮气保护下加入4-[(s)-2-甲基丁氧基]-2,3,5,6-四氟苯乙炔3.0g(11.8 mmol)、对溴碘苯3.34g(11.8mmol)、Pd(PPh3)2Cl2300mg(0.428mmol)和CuI163mg(0.857mmol),随后加入60mL无水Et3N。反应混合液40℃下反应48h。TLC跟踪反应直至完全后,沉淀过滤乙醚萃取,有机相水洗,硫酸镁干燥。真空除掉溶剂,硅胶柱层析得到白色固体4.29g,产率88%。熔点45.5℃。
1HNMR(CCl4,TMS)(δ/ppm):7.40(s,4H),4.04(d,2H,J=6.0Hz),0.82~2.09(m,9H)。19FNMR(CCl4,TFA)(δ/ppm):60.00(d,2F,J=18.8Hz),79.50(d,2F,J=18.8Hz);MS(m/z):416(M+,43.6),414(M+,35.73),346(83.90),344(100.00)。
3.9 1-[(4-正戊氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E1)
在一干燥25mL三颈瓶,氮气保护下加入1-(4-溴苯基)-2-[4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基]乙炔374 mg(0.9 mmol)、4-正戊氧基-2,3,5,6-四氟苯乙炔235 mg(0.9 mmol)、Pd(PPh3)2Cl230 mg(0.043 mmol)和CuI 17 mg(0.089 mmol),随后加入12mL无水Et3N。反应混合液回流8 h。TLC跟踪反应直至完全后,沉淀过滤乙醚萃取,有机相水洗,硫酸镁干燥。真空除掉溶剂,硅胶柱层析得到白色固体192 mg,产率56%。熔点129.0℃。
1H NMR(CCl4,TMS)(δ/ppm):7.45(s,4H),4.11(t,2H,J=5.0Hz),4.00(d,2H,J=5.0Hz),0.62~2.00(m,18H);19F NMR(CCl4,TFA)(δ/ppm):60.03(m,4F),79.75(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 490、1 440、1 390、1 130、985、840、690 cm-1;MS(m/z):594(M+,23.49),524(18.26),454(100.00)。
以下化合物的合成路线与此化合物相似。
3.10 1-[(4-正己氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E2)
产率56%。熔点114.1℃。
1H NMR(CCl4,TMS)(δ/ppm):7.46(s,4H),4.12(t,2H,J=5.0 Hz),4.01(d,2H,J=5.0 Hz),0.65~2.01(m,20H);19FNMR(CCl4,TFA)(δ/ppm):60.05(m,4F),79.77(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 485、1 438、1 390、1 125、982、840、688 cm-1;MS(m/z):608(M+,66.74),538(25.79),454(100.00)。
3.11 1-[(4-正庚氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E3)
产率58%。熔点107.3℃。
1H NMR(CCl4,TMS)(δ/ppm):7.48(s,4H),4.12(t,2H,J=5.0 Hz),4.01(d,2H,J=5.0 Hz),0.67~2.01(m,22H);19FNMR(CCl4,TFA)(δ/ppm):60.04(m,4F),79.76(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 490、1 440、1 390、1 128、982、840、690 cm-1;MS(m/z):622(M+,77.90),552(29.03),454(100.00)。
3.12 1-[(4-正辛氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E4)
产率49%。熔点106.3℃。
1H NMR(CCl4,TMS)(δ/ppm):7.50(s,4H),4.14(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.66~2.00(m,24H);19F NMR(CCl4,TFA)(δ/ppm):60.03(m,4F),79.75(m,4F);IR(KBr):2960、2870、2200、1 520、1 505、1 495、1 441、1 395、1 130、990、840、694 cm-1;MS(m/z):636(M+,24.33),566(11.21),454(100.00)。
3.13 1-[(4-正壬氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E5)
产率49%。熔点99.1℃。
1H NMR(CCl4,TMS)(δ/ppm):7.48(s,4H),4.12(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.68~2.00(m,26H);19FNMR(CCl4,TFA)(δ/ppm):60.04(m,4F),79.77(m,4F);IR(KBr)(cm-1)2 960、2 870、2 200、1 520、1 505、1 495、1 440、1 394、1 130、990、840、694;MS(m/z):650(M+,25.11),580(15.17),454(100.00)。
3.14 1-[(4-正十二氧基-2,3,5,6-四氟苯基)乙炔基]-4-[(4-((s)-2-甲基丁氧基)-2,3,5,6-四氟苯基)乙炔基]苯(E6)
产率56%。熔点:89.2℃。
1H NMR(CCl4,TMS)(δ/ppm):7.49(s,4H),4.10(t,2H,J=5.0 Hz),4.00(d,2H,J=5.0 Hz),0.70~2.00(m,32H);19F NMR(CCl4,TFA)(δ/ppm):60.06(m,4F),79.80(m,4F);IR(KBr):2 960、2 870、2 200、1 520、1 505、1 495、1 440、1 394、1 130、990、840、694cm-1;MS(m/z):692(M+,49.32),622(24.20),454(100.00)。
4 结论
在双苯二乙炔的骨架引入氟原子及四氟亚苯基基元,设计合成了A~E 5个系列的含氟双二苯乙炔液晶,利用Sonogashira交叉偶合反应完成了适于工业化的合成方法。用附带加热台的偏光显微镜及DSC方法,测定了它们的液晶性。
研究发现:1)侧向氟原子取代可以抑制近晶相的出现(A);2)单一的四氟亚苯基有利于抑制近晶相出现,形成温度200℃以上、高清亮点、低熔点液晶化合物,尽管与不含氟母体化合物比较,清亮点有所下降,但是在应用方面是可以接受的(C);3)在化合物C增加对称的2个氟原子,形成的B化合物,液晶性下降,原因是分子间引力进一步减弱;4)在化合物D中有2个四氟亚苯基,分子间引力与化合物C比较,大大减弱,不仅清亮点下降,而且液晶温度区间大大变窄;5)侧键为手性基团的化合物E(见表3),与它的母体化合物D比较,其液晶性几乎遭到破坏。
应用Sonogashira交叉偶合法建立的合成方法,不仅可以合成结构对称的分子,也可以合成结构不对称的分子。可以工业化生产。
化合物A与B是2类介电常数正极性液晶,化合物C是介电常数非极性液晶,它们有高清亮点及足够宽的温度区间。氟取代基的强烈拉电荷作用,使它们避免了非含氟化合物母体的化学不稳定,提高了抗氧化性。由于保留了母体的大的光学双折射性能,进一步降低黏度,并且与上述各种优点结合,有利于制备高性能的液晶混合物,可以用于光子学方面的液晶显示技术。
[1]C SHsu,K F Shyn,Y Y Chang,et al.Synthesis of laterally substituted bistolane liquid crystals[J].Liquid Crystals,2000,27:283-287.
[2]闻建勋,刘克刚,李衡峰.含氟双二苯乙炔类化合物、制备方法及用途:中国,1293180[P].2001-05-02.
[3]Kegang Liu,Hengfeng Li,Kan Wang,et al.Synthesis and characterization of novel fluorinated bistolane-type liquid crystals[J].Liquid Crystals,2001,28:1463-1467.
[4]Yuelian Xu,Yueqing Hu,Qi Chen,et al.Synthesis and characterization of octafluorinated 1,2-(4,4′-dialoxyaryl)acetylene monomers and 1,4-bis[4′,4″-dialkoxyphenyl)ethynyl]benezene dimmers[J].JMater Chem,1995,5:219-221.
[5]CPugh,SK Andersson,V Percec.Phase transfer Pd(0)/Cu(I)catalysedpolymerizationreactions7.Synthesisand thermotropic behaviourof1,4-bis[2-(3′,3″-difluoro-4′,4″-di-n-alkyloxyphenyl)-ethynyl]benzene dimers[J].Liquid Crystals,1991,10:229-232.
[6]JianxunWen,Minquan Tian,QiChen.Novel fluorinated liquid crystals.Part VI.The synthesis and phase transition of novel cholesteric liquid crystals containing 1,4-tetrafluorophenylene units[J].Journal of Fluorine Chemistry,1994,68:117-120.
