Importin α1与HIV-1整合酶相互作用及其对病毒复制的影响
2014-08-11敖竹君KalleshDanappaJayappa姚小剑
敖竹君,Kallesh Danappa Jayappa,姚小剑
(曼尼托巴大学医学院 微生物系人类逆转录病毒实验室,温尼伯 曼尼托巴,加拿大)
Productive infection of non-dividing cells by HIV-1 requires viral double stranded DNA (dsDNA), which associates with viral and cellular proteins in a large preintegration complex (PIC) to actively enter through the intact nuclear membrane for its integration.Such an active, energy-dependent nuclear import process suggests that HIV-1 employs the cellular nuclear import machinery to accomplish its PIC nuclear import during viral replication[1].At the molecular level, the active nuclear import ability of HIV-1 is attributed to the karyophilic properties of viral PICs.It is known that several viral nucleophilic proteins, including integrase (IN), matrix (p17) and Viral protein R (Vpr), are associated with this nucleoprotein complex and play significant roles in HIV-1 nuclear import[2-7]. Moreover, a unique DNA structure, known as the central DNA flap in viral cDNA, has also been implicated in this viral replication step[8-9]. Interestingly, among them, HIV-1 IN and the central DNA flap collectively contribute to HIV-1 nuclear import not only in non-dividing cells but also in dividing cells[8,10-12].It suggests that the nuclear entry of HIV-1 PICs in dividing cells may not be a passive process.Consistently, it was also reported that the nuclear import of HIV-1 PICs may not rely on mitosis in cycling cells[13].
HIV-1 IN, a 32kDa enzyme, is known to be closely associated with the viral PIC during the early stage of HIV infection.Within the PIC, either end of the viral cDNA may be joined through oligomerized IN[14-17]. This DNA-IN interaction may also play an important role for PIC nuclear import and the viral DNA integration.In fact, an earlier study by Gallay and coworkers, suggested for the first time that HIV-1 infection in non-dividing cells may be through the recognition of IN by the host importin/karyopherin pathway[3]. Their study also showed that the nuclear import of viral PIC from a mutated HIV-1(MA(NLS/(Vpr/IN(NLS) was significantly affected.Moreover, a number of subsequent studies have indicated that different IN-fusion proteins, including GST-IN, GFP-IN, PK-IN, HA-IN, and Flag-IN, predominately localized in the nuclei in transfected cells, indicating the karyophilic nature of this protein[11-12, 18-23].
While extensive studies have been carried out on the nuclear import step of HIV-1 replication, the precise molecular mechanism of how HIV-1 IN contributes towards PIC nuclear import is still not fully understood. In particular, it remains to be determined which host nuclear import pathway(s) are employed by HIV IN to ensure active HIV nuclear translocation. The Impα/Impβ-mediated nuclear import pathway is a well characterized pathway for numerous macromolecules, transporting from the cytoplasm to the nuclei in the cells (reviewed in[24-25]. While the Impα subunit harbors a nuclear localization signal (NLS) binding site, the Impβ subunit mediates the interaction with the nuclear pore complex (NPC).Interestingly, Gallay et al have shown that HIV-1 IN is able to interact with the cellular import adapter Rch1 (a 32 N-terminal truncated form of Impα1) in aninvitropull-down assay; furthermore, they found that two IN mutants K186Q and Q214/216L were unable to bind to Rch1[3]. This observation was further confirmed by anotherinvitrobinding study by Hearps et al[26]. However, whether and how HIV-1 IN interacts with Impα1 under the physiological condition and its impact on HIV-1 replication remains to be verified.
In the present study, we have performed a more detailed study to investigate how HIV-1 IN interacts with the Impα/β nuclear import pathway.Our results showed that HIV-1 IN interacts with a full length Impα1. Furthermore, shRNA-mediated Impα1 knockdown in CD4+ T cells led to reduced viral cDNA nuclear import and viral replication, demonstrating a functional role of the IN interaction with the Impα/β pathway during HIV-1 PIC nuclear import and replication.
1 Materials and methods
1.1 Construction of different viral and cellular protein expressors- The human importin α1 (Karyopherin alpha2) expressor, CMV T7-Impα1-myc was purchased from OriGene Technologies. The SVCMVin-IN-YFP and YFP-IN plasmids were previously described[12]. The Matchmaker Chemiluminescent Co-IP vector Set was purchased from Clontech Laboratory. To generate pProLabel-Impα1, the cDNA encoding Impα1 wild type was subcloned into pProLabel-C vector at SalI/BamHI sites. The Moloney murine leukemia virus (MMLV)-based vector pFB-Luc was purchased from Stratagene Inc. The HIV-1 proviral clone pNL4.3-Nef+/GFP+(pNL4.3-GFP) and HIV-1 provirus pNL4.3(Bgl/Luc+were described in previous reports[27-28].
1.2 Antibodies and chemicals-The antibodies used in immunoprecipitation or western blotting were obtained as follows. The purified rabbit anti-GFP polyclonal antibody was obtained from Molecular Probes Inc. The mouse anti-T7 antibody was obtained from Novagen Inc (Darmstsdt, Germany).The goat anti-karyopherin α1 and mouse anti-β-actin antibodies were obtained from Santa Cruz Biotechnology and Abcam Inc. The ECLTMHRP-conjugated sheep anti-mouse IgG was purchased from Amersham Biosciences.The western blot detection ECL kit was purchased from PerkinElmer Life Science (Boston, MA).NP-40 alternative and puromycin were purchased from Calbiochem.The ProLab Detection Kit II was obtained from Clontech.
1.3 Cell culture and transfection-Human embryonic kidney 293T cells and MLV packaging phoenix cells were maintained in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin and streptomycin.The CD4+C8166 T cells were maintained in RPMI-1640 medium containing 10% FBS and antibiotics. DNA transfection in 293T cells was performed with a standard calcium phosphate DNA precipitation method.
1.4 Establishment of the Impα1 knockdown cell line - The pLKO1 lentiviral vector plasmids containing the Impα1 siRNA hairpin, which belong to the TRC-Hs1.0 library, were obtained from Open Biosystems.The hairpin consists of a 21 base stem and 6 base loop.The sense oligo nucleotides sequence was 5’-CTACCTCTGAAGGCTACACTT-3,’ which targeted nucleotides 1661 to 1681 of Impα1. The Impα1 shRNA pseudotyped lentiviral vector particles were produced in 293T cells by co-transfecting pLKO1-shImpα1 vector, the packaging plasmid pCMV(R8.2 and a vesicular stomatitis virus glycoprotein (VSV-G) expression plasmid. The pLKO1 vector plasmid containing scramble shRNA was also obtained from Open Biosystems and used to produce control vector. After 48 h of transfection, the vector particles (VPs) from the supernatant were concentrated by ultracentrifugation (at 32,000rpm) for 1.5 hour at 4℃. To establish cell lines stably expressing shImpα1 or scRNA, 1×106C8166 T cells were transduced with VPs (equivalent to 300 ng of Gag-p24) overnight.After 48 hours, transduced cells were incubated with fresh RPMI containing puromycin (0.5 (μg/mL). After one-week of selection, the cell lines were subjected to western blot to detect the expression of Impα1 and infection experiments.
1.5 IN/Impα1 binding assays using immunoprecipitation (IP) and western blot (WB) -To test protein expression and IN/Impα1 interaction, 293T cells were co-transfected with the corresponding protein expression plasmids.After 48 hours of transfection, cells were washed with PBS and 90% cells were lysed with lysis buffer (199 medium containing 0.25% NP-40 alternative and a protease inhibitor cocktail (Roche)) on ice for 30 min and clarified by centrifugation at 13,000 rpm for 30 min at 4℃.The supernatant was subsequently subjected to IP with a rabbit anti-GFP antibody.Immunoprecipitates were then resolved by 10%-12.5% SDS-PAGE, followed by western blot using corresponding antibodies. To detect the protein expression, 10% of the cells were lysed with 0.5% NP-40/Tris buffer, and directly loaded onto a 10% SDS-PAGE gel, followed by western blot using corresponding antibodies.
1.6 IN/Impα1 binding assays using chemiluminescent Co-IP system-ProLabel is the α fragment of the split β-galactosidase enzyme and can be expressed as a tag on a protein of interest. The prolab-fusion protein alone has no enzymatic activity. When the ProLabel tag combines with thefragment of β-galactosidase, which is supplied in the detection kit, an active enzyme will be reconstituted that will then cleave the chemiluminescent substrate.The resulting signal can be detected and quantified with a standard luminometer.In this study, the wild type Impα1 was cloned into the pProlab-T vector and used for analysis its interaction with YFP-IN or IN-YFP. The procedure for detecting the IN/Impα1 interaction by chemiluminescent Co-IP system was carried out following the manufacturer’s instructions (Clontech). Briefly, after co-transfection of 293T cells with YFP-IN, IN-YFP and pProlab-Impα1, cells were harvested at 48 hours later and lysed by 0.25% NP-40 in 199 medium and followed by IP with rabbit anti-GFP antibody. The immunoprecipitates were resuspended with lysis/complementation buffer, provided in the ProLabel Detection Kit II, and transfer to a well in 96-well plate.Then the substrate mix was added and the chemiluminescent signal was measured with a PolarStar optima microplates reader (BMG Labtech).
1.7 Virus production and infection-Production of pNL4.3-GFP+ and VSV-G-pNL4.3-Luc+virus stock was previously described[27]. Virus titers were quantified using HIV-1 p24 Antigen Capture Assay Kit (purchased from The NCI-Frederick AIDS Vaccine Program).To analyze HIV-1 infection in dividing and non-dividing Impα1-KD-C8166 T cells, the Impα1-KD-cells were pre-treated with 2 μg/ml of aphidicolin (Fisher Scientific) for 24 hours.Aphidicolin treated and mock-treated C8166 cells were then infected with equivalent amounts of VSV-G pseudotyped pNL4.3-Luc+virus (adjusted by virion-associated GagP24 level) for 2 hours.Cells were subsequently washed and incubated with fresh medium. At 48 h post-infection, the infection level was monitored by measuring the luciferase activity (luc) and Gagp24 in the supernatant as described previously[28]. To monitor HIV-1 replication kinetics in Impα1-KD-cells, pNL4.3-GFP+ viruses were used to infect C8166 stable T cell lines and at different time points after infection, viral production levels were monitored by measuring HIV-1 Gag-p24 antigen present in the supernatant using HIV-1 Gag-p24 ELISA.
For production of VSV-G pseudotyped MLV vector particles, a MLV-based retroviral plasmid (pFB-Luc) containing the luciferase gene and a VSV-G plasmid were co-transfected into phoenix cell line that contained thegag-polgene.After 48 hours, supernatants containing MLV particles were filtered through a 0.45 μm filter and used to infect CD4+ C8166 T cells.At 48h post-infection, the level of MLV infection was monitored by measurement of the luc activity.
1.8 Real-time quantitative PCR analysis - HIV pNL4.3-GFP+ were treated with 340I U/ml DNAse (Roche Molecular Biochemicals) for 1 hour at 37oC to remove residual plasmid DNA and then used to infect Impα1-KD- and scRNA-C8166 stable cell lines.At 24 hours post-infection, cells were washed in phosphate-buffered saline and subjected to DNA isolation using a QIAmp blood DNA minikit (Qiagen) following manufacturer’s instructions.The quantity of total HIV-1 DNA, 2-LTR-circles and integrated DNA was estimated using a Mx3000P real-time PCR system (Stratagene, CA). The total HIV-1 DNA content was detected using primers targeting the HIV-1 LTR (HIV-FOR) and Gag genes (HIV-REV)[29]. 2-LTR circles were quantified using primers that amplified LTR-LTR junctions (MH535 and MH536)[30].The extent of integrated HIV-1 cDNA was determined by anAlu-LTR- nested PCR[31].Briefly, in the first round of PCR, the portion of integrated HIV-1 and host cellular genomic DNA were amplified using the primers targeting HIV-1 (M661) andAlu(Alu-HIV) sequence-specific primers.In the second round of PCR, the U5 and R region of HIV-1 from the first round PCR amplicons were quantified using M667 and AA55 primers.All real-time PCR reactions were carried out in 20 μl final volume consisting of 1X FastStart DNA Master SYBR Green I (Roche diagnostics, Germany), 0.2 μm each sense and antisense primer and 2 μl template DNA. The following optimized thermal conditions were used for total HIV-1 DNA and 2-LTR circles: initial hot start (95°C for 15 min) followed by 40 to 50 cycles of denaturation (94°C for 30 s), primer annealing (60°C for 30 s) and extension (72°C for 1 min).The cycling conditions for integrated DNA were adopted from Suzuki et al 2003 without any further modifications.The copy numbers of total DNA, 2-LTR and integrated DNA were calculated in reference to standard curves obtained from amplification of known copies of plasmid DNA having matching DNA sequences and expressed as copy numbers per cell with the amount of starting DNA template normalized by the amplification of the β-globin gene.
2 Results
2.1 The interaction between Impα1 and HIV-1 IN - Previousinvitrobinding studies have shown that IN is capable of interacting with Rch1, a 32 N-terminal truncated form of Impα1, suggesting that the importin/karyopherin pathway can mediate the nuclear translocation of HIV-1 IN[3]. However, whether or not this viral/cellular protein interaction can be detected in the cells, the molecular mechanism of how these viral/cellular proteins interact with each other and its impact on HIV-1 replication are still not fully understood.To gain more insight into the interaction between HIV-1 IN and Impα1, we first used a cell-based co-immunoprecipitation (Co-IP) assay to detect their interaction in 293T cells.YFP, YFP-IN or IN-YFP expressing plasmid was co-transfected with a full-length human Impα1 expressor (CMV-T7-Impα1) in 293T cells, as indicated in Figure 1A.After 48 h, cells were collected and lysed with NP-40 buffer (0.25% NP-40 in 199 medium). The lysates were subsequently subjected to immunoprecipitation (IP) using rabbit anti-GFP antibody and the co-pulled down Impα1 was detected by western blot with an mouse anti-T7 antibody (Fig 1A, upper panel).Results showed that a band corresponding to T7-Impα1 could be detected in the YFP-IN and IN-YFP IP samples, but not in YFP IP sample (Fig 1A, upper panel). This Impα1-IN binding were not due to variation in the expression of either T7- Impα1 or YFP-IN/IN-YFP, suggesting that HIV-1 IN may specifically interact with Impα1 in the cells (Fig 1A, middle or lower panel).
We next examined the IN/Impα1 interaction quantitatively by using a chemiluminescent Co-IP system.Briefly, the Impα1 cDNA was cloned in frame at the C-terminus of a ProLabel-tag protein. When combined with an enzyme acceptor during the reaction, the Prolab-tag formed an active enzyme that was able to cleave the chemiluminescent substrate (as described in Materials and Methods). To quantitatively measure the IN/Impα1 interaction, YFP-IN or IN-YFP were co-transfected with pProlab-Impα1 (PL-Impα1) plasmid into 293T cells (Fig 1B). Meanwhile, YFP was co-transfected with PL-Impα1 vector as a negative control. After 48 hours, cells were lysed and immunoprecipitated with anti-GFP antibody, and the presence of the PL-Impα1 in the IP samples were analyzed by ProLabel Detection Kit II, following manufacturer′s instruction.As expected, we observed a significant PL-activity in samples co-transfected YFP-IN (or IN-YFP) with PL-Impα1, while there was no signal detected in sample transfected YFP and Impα1 (Fig 1B, upper panel). This Impα1-IN binding were not due to variation in the expression of PL-Impα1 in different samples (Fig 1B, lower panel), clearly demonstrating the interaction between IN and Impα1
2.2 Stable knockdown of Impα1 in CD4+ T cell impairs HIV-1 infection - To validate the functional role of Impα1 in HIV-1 replication, we utilized a shRNA-lentiviral vector system to stably knockdown (KD) Impα1 in a CD4+ C8166 T cell line and analyzed the effect of Impα1 depletion on the early stage of HIV-1 infection.After the establishment of Impα1-KD and scRNA stable cell lines, the efficiency of Impα1 knockdown was examined by the western blot (Fig 2A). Concomitantly, the growth kinetics of Impα1-KD cells was assessed in comparison with scRNA-cells. The results show that the Impα1-KD cells displayed slightly slower growth kinetics compared to scRNA-cells (data not shown).
We next tested HIV infection in dividing and non-dividing Impα1-KD- and scRNA-cells.To generate non-dividing cells, cells were treated with alphidicolin (2 μg/ml) for 24 hours prior to viral infection.To test the single cycle HIV replication in Impα1-KD-T cells, equal amounts (1×106cells) of dividing and non-dividing Impα1-KD- and scRNA-cells were infected with VSV-G-pseudotyped pNL4.3Env-/luc+ (10 ng virus-associated p24 antigen). At 48 hours post infection, HIV-1 infection level was monitored by measuring luciferase activity.The results showed that in both dividing and non-dividing Impα1-KD-C8166 cells, viral infectivity was reduced by approximately 50%-70%, as compared with that in scRNA-cells (Fig 2B, upper panel). Similar levels of reduced luciferase activity were also obtained in Impα1-KD-HeLa cells (data not shown). HIV-1 p24 antigen levels were measured from the supernatant of each infected cultures by anti-p24 ELISA. A significantly reduced HIV p24 production was observed in both dividing and non-dividing Impα1-KD-cells (Fig2B, lower panel). This result indicates that Impα1 knockdown attenuated HIV-1 infection in both dividing and non-dividing C8166 T cells.

A) Full-length human T7-Impα1 plasmid was co-transfected with YFP, YFP-IN or IN-YFP plasmids into 293T cells. After 48 h of transfection, cell lysates were immunoprecipitated with rabbit anti-GFP antibody. The immunoprecipitates were resolved using 10% SDS-PAGE and the bound T7-Impα1 was detected by WB with an anti-T7 antibody (upper panel).To check protein expression, 1/10th transfected cells were probed with anti-T7(middle panel) or anti-GFP antibody (lower panel). B) YFP, YFP-IN or IN-YFP was co-transfected with PL-Impα1 in 293T cells. After 48h of transfection, cells were lysed and immunoprecipitated with anti-GFP antibodies and the chemiluminescent signals from PL-Impα1 present in the complexes were measured by using ProLabel Detection Kit II and valued as relative luminescence units (upper panel). The lower panel is the PL activity of PL-Impα1 in cell lysates. Results are representative of three independent experiments.Fig 1 Impα1 binds to HIV-1 IN
In parallel, we tested VSV-G pseudotyped MLV-vector infection in these cell lines. Since a luciferase gene was inserted in the MLV vector, the MLV-vector infection levels were also monitored by the measurement of the luciferase activity at 48 hours post-infection. Results show that the MLV infection in dividing Impα1-KD-cells was reduced approximately 30% of that in scRNA-cells.As expected, in non-dividing Impα1-KD- and scRNA-cells, MLV could not initiate infection (Fig 2C).These results indicate that the Impα1-KD in dividing C8166 cells also modestly inhibited MLV infection.
2.3 Effect of Impα1-KD on early steps of HIV-1 infection and its impact on viral replication -The above experiments demonstrated that Impα1 depletion in C8166 T cells impaired HIV-1 infection.To pinpoint at which step(s) the viral infection was affected in Impα1-KD-cells, cells were infected with DNAse-treated VSV-G pseudotyped viruses. After 24 hours of infection, cells were harvested and DNAs were extracted to quantify total late reverse transcripts, 2-LTR circles and the integrated provirus using RQ-PCR.The results showed that the viral cDNA synthesis was not affected in Impα1-KD-cells (Fig 3A).However, over a 50% reduction of 2-LTR circles and integrated proviral DNA levels was observed in HIV infected Impα1-KD-cells (Fig 3B、 C).This indicates that HIV infection was inhibited specifically at the nuclear import step in Impα1-KD C8166 T cells.

A) CD4+ C8166 T cells were transduced with lentiviral vector particles containing Impα1 shRNA or scRNA.After 2 days of transduction, cells were selected with puromycin for one week. Then, each cell line was subjected to WB to verify Impα1 expression by using anti-Impα1 antibody (Upper panel).The β-actin expression was checked as control (lower panel). B) The nondividing (aphidicolin-treated, right) or dividing Impα1-KD- and scRNA-C8166 cells were infected with equal amounts of VSV-G pseudotyped HIV-1 (pNL4.3VSV-G/luc+).48 hours post infection, equal amounts of cells were collected and the luciferase activity was measured (Middle panel). The HIV Gag-p24 levels in the supernatant were also measured by p24 ELISA (Upper panel). C) The Impα1-KD- and scRNA-cells were infected by a VSV-G pseudotyped MLV-vector containing the luciferase gene, and the luciferase activity of infected cells was checked at 48 hours post-infection (lower panel). Results are representative of three independent experiments. Fig 2 Impα1 knockdown in CD4+T cells impairs HIV-1 infection
To gain more insight into the effect of Impα1 on HIV-1 replication, we next evaluated the kinetics of HIV-1 replication in Impα1-KD- and scRNA-cells by infecting cells with equal amounts of pNL-4.3-GFP virus (100 pg Gagp24/sample)[27]. At different time points, supernatants were collected, and the viral replications were monitored by Gag-p24 ELISA.Results show that HIV-1 replication kinetics progressed much faster in scRNA-cells than in Impα1-KD-cells (Fig 3D).Viral replication peaked at day 8 and declined at day 10 due to extensive virus-induced cytopathic effect, including syncytia formation and cell death.However, viral replication in Impα1-KD-cells progressed significantly slower. At days 4 and 6, there was approximately 4-folds decrease of Gagp24 production in Impα1-KD-cells as compared to viral infection in scRNA-cells.The extent of the inhibitory effect was more significant than in single cycle infection.This could be due to an accumulative effect by multiple cycles of viral infection in C8166 cells.We could not continue our investigation after 8 days of infection, because of an extensive syncytia formation and cell death in scRNA-cells.Overall, these results clearly indicate that the presence of Impα1 significantly contributes to an efficient HIV-1 replication and spread in C8166 T cells.
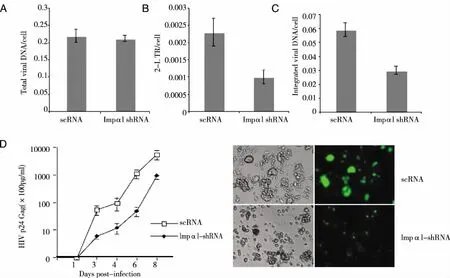
3×106 Impα1-KD- and scRNA-C8166 cell lines were infected with DNase-pretreated HIV-1 pNL4.3-GFP virus for 2 hours, washed twice with medium and cultured for 24 hours.Cells were harvested and viral DNAs from infected cells were isolated and HIV-1 late reverse transcription products (A), HIV-1 2-LTR circles (B), and the integrated HIV-1 DNA (C) were analyzed by RQ-PCR using corresponding primers as described in Materials and Methods.D) Inhibitory effect of Impα1 depletion on HIV-1 multiple cycle replication and progression. 1x106 Impα1-KD- and scRNA-C8166 cell lines were infected with HIV-1 PNL 4.3 GFP+. At different time points, as indicated, the supernatant was collected and HIV Gag-p24 level was measured to monitor virus replication level (Left panel). Right panel show the GFP expression of infected cells at day 6 after infection.Results are representative of two independent experiments.Fig 3 Inhibitory effect of Impα1 knockdown on HIV-1 nuclear import and replication
3 Discussion
One important question that remains regarding HIV IN nuclear is the identity of the host nuclear import cofactors that are hijacked by HIV IN.The first host cofactor that has been shown to interact with IN in aninvitrobinding assay was a N-terminal 32-amino acid truncated form of Impα1 (Rch1)[3].However, although a later studies confirmed IN/Impα1 binding usinginvitroassays[26], stronger evidence is needed to illustrate this viral/cellular interaction and its functional role in HIV nuclear import and replication. In this study, we performed cell-based Co-IP experiments to further investigate the IN/Impα1 interaction.Interestingly, our data revealed that HIV-1 IN was able to bind to Impα1 in the cells (Fig 1).
To further investigate whether the IN/Imp( interaction contributes to HIV-1 replication, we specifically knocked down Impα1 expression in CD4+ C8166 T cells and tested its impact on HIV replication.Interestingly, results showed that depletion of Impα1 led to an approximately 50%-70% reduction in single cycle replication of HIV-1 in dividing and non-dividing cells as compared to the control cells (Fig 2C).Real-time PCR analysis revealed that Impα1 depletion did not affect viral reverse transcription, but resulted in approximately a 50% decrease in 2-LTR DNA and integrated DNA (Fig 3A-C).Since the reduced level of integrated viral DNA was comparable to the decreased level of 2-LTR DNA, it suggests that the attenuation of viral replication occurred mainly at the nuclear import step.Furthermore, the nuclear entry of HIV-1 PICs in dividing cells may also not be a passive process since the inhibitory effect mediated by Impα1-depletion could also been observed in dividing cells (Fig 2). Consistently, similar observations in dividing cells were also documented for Imp7[28]and for TRN-SR2[32].Indeed, studies by Katz et al. clearly showed that an efficient HIV-1 and RSV nuclear entry can occur independently of mitotic nuclear disassembly in cycling cells[13, 33]. It is possible that during the early stage of HIV-1 infection, IN interacts with different nuclear import factors and facilitates the transport of HIV-1 PIC through nuclear pore, and that this process is also required for post-nuclear processing.
VSV-G pseudotyped MLV vector (VSV-G-MLV) infection was also evaluated in the study and as expected, MLV vector infection could only infect dividing C8166 cells.Interestingly, a modest reduction (approximately 30%) of MLV infection was seen in Impα1-KD cells (Fig 2C). At this point, we still do not know the mechanism behind this phenotype. It could be possible that Impα1 is also required for efficient MLV infection. Interestingly, a previous study by Katz et al. indicated that MLV may not be strictly dependent on mitosis for nuclear entry in cycling cells[13]. Also, it was reported that nondividing human macrophages could be successfully transduced with the FMLV-derived vector[34]. Whether Impα1 could also bind to MLV integrase or other viral proteins, such as the capsid protein, and how Impα1 contributes to MLV infection requires further investigation.However, the results clearly indicated that the impact of Impα1-depletion on HIV-1 infection was more significant than MLV infection, demonstrating that Impα1 plays a more prominent role in HIV-1 infection.
Six Impα family isoforms (α1/Rch1, α3/Qip1,α4,α5, Impα6, and Impα7) have been identified in human cells[35-40]. These six isoforms share about 50% sequence identity overall and are grouped into three subfamilies.Invitrostudies indicate that while that individual human Impα are able to import the same target protein, each isoform differs in its binding efficiency[35, 37]. Recently, by using co-IP method, we have demonstrated that HIV IN specifically interacts with Impα3 and contributes to HIV-1 nuclear import and replication in dividing and non-dividing cells[41]. In this study, we found that HIV IN also interacts with Impα1 and knockdown Impα1 in CD4+ C8166 cells lead to about 50% reduction of viral DNA nuclear import and viral replication. Even though the effect of Impα1 in HIV-1 nuclear import is weak than of Impα3[41], our study strongly suggest that HIV-1 IN could alternatively bind to Imp α1 such as in the absence of Impα3, or HIV IN can hijack different Impα in different cell types for an efficient viral replication. Therefore, further investigation on how HIV IN hijack both Imp α1 and Impα3 for an efficient viral replication will facilitate a better understanding of HIV-1 nuclear import.
Acknowledgements:X-J. Y is a recipient of the Basic Science Career Development Research Award from The Manitoba Medical Service Foundation (MMSF).This work was supported by CIHR grants (HOP-63013 and HOP-81180)and the Leaders Opportunity Fund Award from Canadian Foundation of Innovation (CFI) to X-J. Y.
[References]
[1] Bukrinsky M, I.N. S, Dempsey M P, Stanwick T L, et al. Active nuclear import of human immunodeficiency virus type 1 preintegration complex[J].Proc Natl Acad Sci USA, 1992, 89(14):6580-6584.
[2] Bukrinsky M, Haggerty S, Dempsey M P, et al. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells[J].Nature,1993, 365(6447):666-669.
[3] Gallay P, Hope T, Chin D, et al. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway[J].Proc Natl Acad Sci U S A,1997, 94(18):9825-9830.
[4] Gallay P, Swingler S, Aiken C, et al. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator[J].Cell,1995, 80(13):379-388.
[5] Heinzinger N K, Bukinsky M I, Haggerty S A, et al. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells[J]. Proc Natl Acad Sci U S A,1994, 91(15):7311-7315.
[6] Vodicka M A, Koepp D M, Silver P A, et al. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection[J]. Genes Dev,1998, 12(2):175-185.
[7] Nie Z, Bergeron D, Subbramanian R A,et al. The putative alpha helix 2 of human immunodeficiency virus type 1 Vpr contains a determinant which is responsible for the nuclear translocation of proviral DNA in growth-arrested cells[J]. J Virol,1998, 72(5):4104-4115.
[8] Zennou V, Petit C, Guetard D, et al. HIV-1 Genome nuclear import is mediated by a central DNA flap[J]. Cell,2000, 101(2):173-185.
[9] Ao Z, Yao X, Cohen E A.Assessment of the role of the central DNA flap in human immunodeficiency virus type 1 replication by using a single-cycle replication system[J]. J Virol,2004, 78(6):3170-3177.
[10] Sirven A, Pflumio F, Zennou V, et al. The human immunodeficiency virus type-1 central DNA flap is a crucial determinant for lentiviral vector nuclear import and gene transduction of human hematopoietic stem cells[J]. Blood,2000, 96(13):4103-4110.
[11] Bouyac-Bertoia M, Dvorin J D, Fouchier R A,et al. HIV-1 infection requires a functional integrase NLS[J]. Molecular Cell,2001, 7(5):1025-1035.
[12] Ao Z, Fowke K R, Cohen E A, et al. Contribution of the C-terminal tri-lysine regions of human immunodeficiency virus type 1 integrase for efficient reverse transcription and viral DNA nuclear import[J]. Retrovirology,2005, 2(1):62.
[13] Katz R A, Greger J G, Boimel P,et al. Human immunodeficiency virus type 1 DNA nuclear import and integration are mitosis independent in cycling cells[J]. J Virol,2003,77(24):13412-13417.
[14] Miller M D,Farnet C M, Bushman F D.Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition[J]. J Virol,1997, 71(7):5382-5390.
[15] Wei S Q, Mizuuchi K, Craigie R. A large nucleoprotein assembly at the ends of the viral DNA mediates retroviral DNA integration[J]. Embo J,1997, 16(24):7511-7520.
[16] Ellison V, Brown P O. A stable complex between integrase and viral DNA ends mediates human immunodeficiency virus integration in vitro[J]. Proc Natl Acad Sci U S A,1994,91(15):7316-7320.
[17] Farnet C M, Bushman F D. HIV-1 cDNA integration: requirement of HMG I(Y) protein for function of preintegration complexes in vitro[J]. Cell,1997, 88(4):483-492.
[18] Depienne C, Mousnier A, Leh H,et al. Characterization of the nuclear import pathway for HIV-1 integrase[J]. J Biol Chem,2001, 276(21):18192-18107.
[19] Limon A, Devroe E, Lu R, et al. Nuclear localization of human immunodeficiency virus type 1 preintegration complexes (PICs): V165A and R166A are pleiotropic integrase mutants primarily defective for integration, not PIC nuclear import[J]. J Virol,2002, 76(21):10598-10607.
[20] Lu R, Limon A, Devroe E, et al. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication[J]. J Virol,2004, 78(23):12735-12746.
[21] Petit C, Schwartz O, Mammano F. The karyophilic properties of human immunodeficiency virus type 1 integrase are not required for nuclear import of proviral DNA[J]. J Virol,2000,74(15):7119-7126.
[22] Pluymers W, Cherepanov P, Schols D, et al. Nuclear localization of human immunodeficiency virus type 1 integrase expressed as a fusion protein with green fluorescent protein[J]. Virology,1999,258(2):327-332.
[23] Tsurutani N, Kubo M, Maeda Y, et al. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells[J]. J Virol,2000, 74(10):4795-4806.
[24] Lange A, Mills R E, Lange C J, et al. Classical nuclear localization signals: definition, function, and interaction with importin alpha[J]. J Biol Chem,2007, 282(8):5101-5105.
[25] Goldfarb D S, Corbett A H, Mason D A,et al. Importin alpha: a multipurpose nuclear-transport receptor[J]. Trends Cell Biol,2004, 14(9):505-514.
[26] Hearps A C, Jans D A.HIV-1 integrase is capable of targeting DNA to the nucleus via an importin alpha/beta-dependent mechanism[J]. Biochem J,2006, 398(3):475-484.
[27] Ao Z, Yu Z, Wang L,et al. Vpr14-88-Apobec3G fusion protein is efficiently incorporated into Vif-positive HIV-1 particles and inhibits viral infection[J]. Plos one,2008, 3(4):e1995.
[28] Ao Z, Huang G, Yao H, et al. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication[J]. J Biol Chem,2007, 282(18):13456-13467.
[29] Malnati M S, Scarlatti G, Gatto F, et al. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load[J]. Nat Protoc,2008, 3(7):1240-1248.
[30] Butler S L, Johnson E P, Bushman F D. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles[J]. J Virol,2002,76(8):3739-3747.
[31] Suzuki Y, Misawa N, Sato C, et al. Quantitative analysis of human immunodeficiency virus type 1 DNA dynamics by real-time PCR: integration efficiency in stimulated and unstimulated peripheral blood mononuclear cells[J]. Virus Genes,2003, 27(2):177-188.
[32] Christ F. Transportin-SR2 imports HIV into the nucleus[J]. Current Biology,2008,18(16):1192-1202.
[33] Katz R A, Greger J G,Darby K,et al. Transduction of interphase cells by avian sarcoma virus[J]. J Virol,2002, 76(11):5422-5434.
[34] Jarrosson-Wuilleme L, Goujon C, Bernaud J, et al. Transduction of nondividing human macrophages with gammaretrovirus-derived vectors[J]. J Virol,2006, 80(3):1152-1159.
[35] Kohler M, Speck C, Christiansen M, et al. Evidence for distinct substrate specificities of importin alpha family members in nuclear protein import[J]. Mol Cell Biol,1999,19(11):7782-7791.
[36] Cortes P, Zheng-Sheng Y, Baltimore D. RAG-1 interacts with the repeated amino acid motif of the human homologue if the yeast protein SRP1[J]. Proc Natl Acad Sci USA,1994, 91(16):7633-7637.
[37] Kohler M, Gorlich D, Hartmann E, et al. Adenoviral E1A protein nuclear import is preferentially mediated by importin alpha3 in vitro[J]. Virology,2001, 289(2):186-191.
[38] Seki T, Tada S, Katada T, et al. Cloning of a cDNA encoding a novel importin-alpha homologue, Qip1: discrimination of Qip1 and Rch1 from hSrp1 by their ability to interact with DNA helicase Q1/RecQL[J]. Biochem Biophys Res Commun,1997, 234(1):48-53.
[39] Weis K, Mattaj I W, Lamond A I. Identification of hSRP1 alpha as a functional receptor for nuclear localization sequences[J]. Science,1995, 268(5213):1049-1053.
[40] Weis K, Ryder U, Lamond A I. The conserved amino-terminal domain of hSRP1 alpha is essential for nuclear protein import[J]. EMBO J,1996, 15(8):1818-1825.
[41] Ao Z, Danappa Jayappa K, Wang B, et al. Importin alpha3 interacts with HIV-1 integrase and contributes to HIV-1 nuclear import and replication[J]. J Virol,2010, 84(17):8650-8663.
