AMPK/mTOR信号通路在DIO与DR大鼠胰腺中的表达
2014-08-09杜建颖王春妹王启明田德润
韩 洁, 杨 慧,杜建颖, 王春妹, 王启明 ,田德润
(天津医科大学 解剖与组织胚胎学系,天津 300070)
mTOR(mammalian target of rapamycin),即哺乳动物雷帕霉素靶点,在所有的真核生物中普遍高表达,是调节细胞生长的中心,与人类的健康息息相关,许多慢性疾病的发生都与mTOR通路信号的改变有关,如II型糖尿病、肥胖、代谢综合征以及多种肿瘤性疾病[1-3]。mTOR存在于两种复合体中,mTORC1和mTORC2[4]。其中,mTORC1由四个亚基组成,其下游底物包括4E-BP1和S6K等;而mTORC2由五个亚基组成,包括其下游底物包括AKT、PKC等。两种复合体中,mTORC1与代谢联系紧密[5],它可以控制细胞生长,血管发生和代谢等[6- 7]。mTOR通路上游有多种调节因素,如:瘦素(leptin)、脂联素、胃饥饿素(ghrelin)、胆囊收缩素、胰岛素等,这些因素可以在下丘脑和外周器官通过mTOR通路相互影响以调控机体的能量平衡[8]。下丘脑是机体能量调节的中枢,而胰腺作为一个重要的外周器官,对能量平衡和代谢也有重要的影响。我们前期的实验发现食源性肥胖(DIO)大鼠下丘脑AMPK表达下调,导致mTOR通路活性增加。本文旨在比较DIO大鼠和DR大鼠胰腺中mTOR通路的变化,并探讨这种变化对整个机体能量和代谢过程中的作用。
1 材料与方法
1.1 实验动物 100只刚断乳雄性SD大鼠,体重40~50 g。动物饲养于天津医科大学实验动物中心,动物室温度:(22±2)℃;每天光照:7:00~19:00;随意取食饲料和自来水。基础维持饲料适应喂养1周后分组:①高脂饮食组(n=80),给予自制高脂饮食饲料;②对照组(n=20),继续给予基础维持饲料。继续喂养14周后,高脂饮食组体重超过对照组最大体重者,设为肥胖组(DIO);体重低于对照组平均体重的设为肥胖抵抗组(DR)。
1.2 动物饲料 基础维持饲料能量成份:5%脂肪,55%碳水化合物,22%蛋白质,7%灰分,5%纤维素。饲料热量为3.80 kcal/g。
自制高能饲料配方为:50%标准动物饲料,15%猪油,10%白糖,5%奶粉,10%鸡蛋,5%芝麻油,5%花生。能量成份为:30%脂肪,40%碳水化合物,15.5%蛋白质,4%灰分,3%纤维素。饲料热量为4.76 kcal/g。
1.3 取材 各组动物经水合氯醛(500 mg/kg)麻醉后,主动脉插管,用生理盐水快速灌注冲出血液后,用4%多聚甲醛缓慢灌注,至全身肌肉抽搐后停止。迅速剪开腹腔,取胰腺,放入4 ℃4%多聚甲醛溶液中后固定,过夜。
1.4 包埋及切片 部分组织梯度蔗糖脱水(至组织下沉)后,用OCT胶包埋。冰冻切片厚度:6~8 μm。其余组织蒸馏水冲洗后,经梯度酒精脱水、二甲苯透明,石蜡包埋。石蜡切片厚度:4 μm。
1.5 免疫组织化学和免疫荧光
1.5.1 免疫组织化学 石蜡切片经二甲苯和梯度酒精脱蜡至水,0.3% H2O2甲醇溶液灭活过氧化物酶,98℃柠檬酸钠修复抗原,10%山羊血清封闭后,加入一抗(AMPK、4E-BP1),4℃过夜,再经SP试剂盒(二抗和辣根过氧化物酶),DAB显色,苏木素染色,酒精梯度脱水,二甲苯透明,最后用中性树胶封片。PBS替代一抗作为阴性对照。
1.5.2 免疫荧光 冰冻切片经室温复温后水化,经1%Triton-X100增加通透性,10%山羊血清封闭后,加入一抗(S6K),4℃过夜,再避光加入二抗、DAPI,最后用甘油封片。PBS替代一抗作为阴性对照。用倒置荧光显微镜(Olympus IX71)观察并照相;通过比较荧光信号的强弱和荧光细胞的数量对结果进行分析。
1.6 图像分析 针对各组的免疫组化染色结果,分别选取5个独立的视野用Image-Pro Plus 6.0图像分析系统测量出单位光密度值,并求得平均光密度值(OD值)再进行比较分析。
2 结果
2.1 动物模型结果 初始体重高脂饮食组(49.95±2.56)g,对照组(48.65±2.94)g,两组无差别。给予不同饲料4周后开始出现差别,持续到14周。两组最终体重分别为高脂饮食组(545.98±49.49)g,对照组(497.55±37.38)g,具有显著差异(P<0.001)(见图1)。其中,高脂饮食组36只大鼠体重超过对照组最高体重,设置成DIO组(587.56±39.32)g,11只鼠体重低于对照组平均体重,设置成DR组(476.90±15.06)g,对照组体重取中间10只,设置成CO组(503.03±17.24)g。DIO组与DR组、DIO组与CO组之间体重有统计意义(P<0.001),DR组和CO组之间体重无统计意义(见图2)。

注:﹡:P<0.05,﹡﹡:P<0.01,﹡﹡﹡:P<0.001,VS对照组。图1 大鼠体重变化图
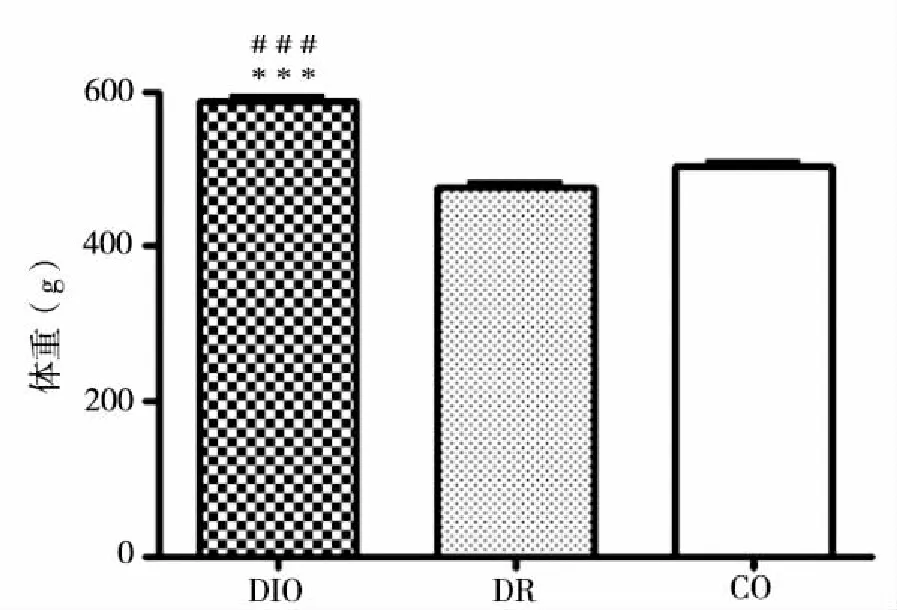
注:﹡﹡﹡:P<0.001,vs DR;﹟﹟﹟:P<0.001,vsCO。图2 分组后大鼠体重图
2.2 免疫组织化学和免疫荧光结果 AMPK在胰腺中的阳性表达以胞浆为主,呈棕黄色颗粒状;以内分泌部细胞表达明显。DIO组明显高于DR组,有统计意义(P<0.05)(见表1、图3)。
表1大鼠胰腺各组免疫组化结果平均光密度值(OD值)比较
注:﹡:P<0.05,vs DIO。
4E-BP1阳性表达同样以胞浆为主,呈棕黄色颗粒状,表达的区域以胰腺的外分泌部细胞最为明显,胰岛外周部亦有表达,DIO组明显低于DR组,有统计学意义(P<0.05)(见表1、图4)。
S6K的免疫荧光阳性表达着色以胞浆为主,呈绿色荧光,胰腺的胰岛外周部有典型的绿色荧光信号,DIO组明显低于DR组(见图5)。
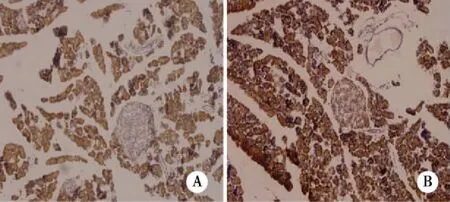
注: A:DIO胰腺;B:DR胰腺。图4 4E-BP1免疫组织化学染色SP法×100

注: A:DIO胰腺;B:DR胰腺。图5 S6K免疫荧光×200
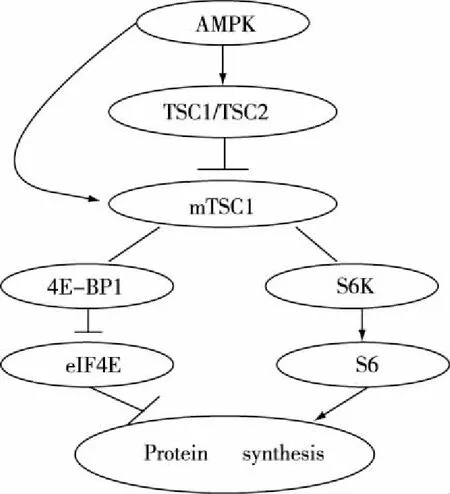
图6 AMPK/mTOR通路图
3 讨论
肥胖及其相关性代谢性疾病在世界的流行越来越受到人们的关注。食源性肥胖大鼠模型的建立过程,模拟了人类肥胖发生的过程。在建模过程中,原本体重无差异的大鼠,部分表现出摄食过度,成为DIO大鼠,另外的部分则维持了相对正常的体重,成为DR大鼠,这表明肥胖这种疾病的发生受基因和环境(如饮食)共同控制,而不是仅受基因或者环境单方面的影响。
果蝇和哺乳动物基因研究表明TSC1/TSC2是位于mTORC1上游的重要抑制剂,磷酸化TSC1/TSC2可以调节mTORC1的活性,缺失TSC1/TSC2会导致mTORC1过度活跃[9]。 AMPK可以磷酸化TSC2保守的丝氨酸位点,抑制mTORC1复合体的活性[10]。而当缺乏TSC2的细胞时给予AMPK后,细胞仍然可以变现出部分抑制,说明AMPK还可以通过除TSC2之外的其他途径直接或间接影响mTORC1活性[11],如AMPK可以通过直接磷酸化raptor(mTORC1复合体的一个亚基)来抑制mTOR通路的活性[12-13]。
AMPK是一种AMP相关的高度保守的异源三聚体丝氨酸/苏氨酸激酶复合体。AMPK可以作为机体能量的开关,在能量不足的情况下被激活[9]。当能量不足时,AMP可以直接绑定AMPKγ亚基的CBS区域,抑制α亚基的丝氨酸的去磷酸化,而这种丝氨酸的磷酸化是AMPK激活所必须的[14]。AMPK可以对中枢神经系统和外周组织中的激素和营养的信号作出反应,进而调节食物摄入和能量消耗。其下游靶点mTOR通路,可以作为细胞内的营养传感器来调节蛋白质合成、细胞生长和代谢。
mTOR通路经典的下游靶标4E-BP1和S6K都有一个特殊的基序(TOS基序),可以使二者直接绑定到mTORC1复合体的raptor上[15]。4E-BP1多个位点磷酸化,使得4E-BP1从eIF4E上解离,并促进eIF4G与eIF4E绑定,最终形成翻译起始复合物。同时,S6K1的389位苏氨酸磷酸化,激活S6K1,随后磷酸化核糖体蛋白S6,最终促进蛋白质的合成。另外,S6K1也可以对准eIF4B的422位丝氨酸,促进其与eIF4A绑定,提高eIF4A解链酶活性,然后启动蛋白质翻译过程[16](见图6)。
胰腺作为机体重要的内分泌器官,分为外分泌部和内分泌部。其中内分泌部起主要作用的是胰岛细胞,包括α细胞、β细胞、D细胞和PP细胞等。α细胞主要位于胰岛的外周部分,分泌胰高血糖素;β细胞和D细胞主要位于胰岛的中间部分,分别分泌胰岛素和生长激素;而PP细胞含量很少,分泌胰多肽。
我们的结果显示,DR大鼠胰岛的周围部分4E-BP1和S6K表达增加,也就是mTOR通路活性与DIO相比明显增加,而其上游AMPK信号却相对减弱。而位于胰岛外周的是胰岛α细胞[17],信号活化增加了胰高血糖素的分泌。胰高血糖素可以激活肝细胞的磷酸化酶,加速糖原分解。也可激活脂肪酶,促进脂肪分解,并且加强脂肪酸氧化,使酮体生成增多。还可以促进氨基酸进入肝细胞。酮体和氨基酸的增加可以促进糖异生,可以与分解的肝糖原共同升高血糖。升高的血糖可以通过下丘脑的AMPK传递饱食的营养信号,刺激下丘脑弓状核分泌抑制食欲的POMC[18],导致食物的摄入的减少,从而使DR大鼠维持较正常体重。此外,胰高血糖素在肝脏促进脂肪分解,也可以从另一方面维持DR大鼠体重。有文献报道,胰岛α细胞缺失的胰岛对葡萄糖刺激产生的胰岛素明显降低[19],也就是说,胰高血糖素刺激了胰岛素的分泌,缺乏其刺激作用将减少胰岛素的分泌,这可能是DR大鼠能维持正常体重的又一重要原因。但确切的机制需要进一步的研究证实。
[参考文献]
[1] Nawroth R, Stellwagen F, Schulz W A, et al. S6K1 and 4E-BP1 are independent regulated and control cellular growth in bladder cancer[J]. PLoS One, 2011,6(11):e27509.
[2] Din F V, Valanciute A, Houde V P, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells[J]. Gastroenterology, 2012,142(7):1504-1515.
[3] Weijenberg M P, Hughes L A, Bours M J, et al. The mTOR Pathway and the Role of Energy Balance Throughout Life in Colorectal Cancer Etiology and Prognosis: Unravelling Mechanisms Through a Multidimensional Molecular Epidemiologic Approach[J]. Curr Nutr Rep, 2013,2(1):19-26.
[4] Wullschleger S,Loewith R, Hall M N. TOR signaling in growth and metabolism[J]. Cell, 2006,124(3):471-484.
[5] Zoncu R, Efeyan A, Sabatini D M. mTOR: from growth signal integration to cancer, diabetes and ageing[J]. Nat Rev Mol Cell Biol, 2011,12(1):21-35.
[6] Garcia-Martinez J M, Alessi D R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1)[J]. Biochem J, 2008,416(3):375-385.
[7] Guertin D A, Sabatini D M. Defining the role of mTOR in cancer[J]. Cancer Cell, 2007,12(1):9-22.
[8] Mannaa M, Krämer S, Boschmann M, et al. mTOR and regulation of energy homeostasis in humans[J]. J Mol Med (Berl), 2013,91(10):1167-1175.
[9] Shaw R J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth[J]. Acta Physiol (Oxf), 2009,196(1):65-80.
[10] Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth[J]. Cell, 2006,126(5):955-968.
[11] Hahn-Windgassen A, Nogueira V, Chen C C, et al. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity[J]. J Biol Chem, 2005,280(37):32081-32089.
[12] Inoki K, Zhu T, Guan K L. TSC2 mediates cellular energy response to control cell growth and survival[J]. Cell, 2003,115(5):577-590.
[13] Gwinn D M, Shackelford D B, Egan D F, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint[J]. Mol Cell, 2008,30(2):214-226.
[14] Sanders M J, Grondin P O, Hegarty B D, et al. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade[J]. Biochem J, 2007,403(1):139-148.
[15] Holz M K, Ballif B A,Gygi S P, et al. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events[J]. Cell, 2005,123(4):569-580.
[16] Liu Y, Vertommen D, Rider M H, et al. Mammalian target of rapamycin-independent S6K1 and 4E-BP1 phosphorylation during contraction in rat skeletal muscle[J]. Cell Signal,2013,25(9):1877-1886.
[17] Liu Z, Kim W, Chen Z, et al. Insulin and glucagon regulate pancreatic alpha-cell proliferation[J]. PLoS One, 2011,6(1):e16096.
[18] Zhan C, Zhou J, Feng Q, et al. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively[J]. J Neurosci, 2013,33(8):3624-3632.
[19] 李光伟,叶丽亚,李静,等.小鼠胰岛α细胞的缺失对移植胰岛功能的影响[J].中华医学杂志, 2002,82(20):1427-1431.
