鱼腥藻PCC7120 α质粒上的parD/E同源基因all7155/asl7156的克隆及表达
2014-08-06陈思礼刘炳君
陈思礼, 张 磊, 刘炳君
(中南民族大学 生命科学学院, 武汉 430074)
毒素-抗毒素系统最初是在大肠杆菌的低拷贝质粒中发现[1],此系统也是引发细胞程序性死亡(PCD)的一个重要途径[2,8].本文中T-A系统基因对是位于鱼腥藻PCC7120的α质粒上的一对基因,通过NCBI中BLAST比对后发现目的基因编码的蛋白质与大肠杆菌中的ParD/E毒素-抗毒素基因对编码蛋白同源, 而ParD/E毒素抗毒素系统是R1质粒上发现的分别编码毒素蛋白Kid和抗毒素蛋白Kis的基因对[3,4],此毒素-抗毒素基因对在E.coli的R1质粒上被发现[3,4],且ParE毒素基因与F质粒上的Ccd毒素抗毒素系统中的CcdB毒素基因具有高度同源性[5,6],ParE和CcdB编码蛋白的作用目标均为DNA螺旋酶,在DNA的复制阶段破坏DNA双链起到其毒素蛋白的作用[5],故推断其编码的蛋白具有与毒素-抗毒素作用相似的活性功能.并以此作为本文的理论基础.
本文分别克隆了all7155和asl7156基因,再将目的基因片段插入已双酶切的带有组氨酸标签的表达载体PET30a(+)中,用以构建表达载体,并加入IPTG进行目的蛋白诱导表达,通过SDS-PAGE电泳初步检测.为进一步研究all7155/asl7156的毒素抗毒素基因对的相互作用提供了一定的研究基础.
1 材料与方法
1.1 材料与试剂
鱼腥藻sp.PCC7120(中国科学院水生物研究所),pMD18-T Vector(Takara公司),E.coliDH5α和E.coliBL21(DE3),表达载体pET30a(+)为本实验室保存.DNA聚合酶(Fermentas公司),限制性内切酶、T4 DNA连接酶(Takara公司),琼脂糖凝胶回收试剂盒及质粒DNA提取试剂盒(Axygene产品),PCR引物(南京金斯瑞生物科技有限公司),克隆片段测序(南京金斯瑞生物科技有限公司测序确定).
1.2 分子生物学操作
参照分子克隆实验指南[10]提取PCC7120质粒,以其为模板,以all7155-F和all7155-R为引物(见表1), PCR扩增all7155基因片段;以asl7156-F和asl7156-R为引物(见表1),PCR扩增asl7156基因序列片段.引物中添加BamH I和Hind III酶切位点,PCR扩增产物纯化后与pMD18-T Vector载体连接,转化E.coliDH5α,蓝白斑筛选阳性克隆酶切鉴定测序正确后保存质粒,分别命名为pMD18-T-7155和pMD18-T-7156.

表1 PCR引物序列
1.3 构建重组表达载体
利用BamH I和Hind III双酶切质粒pET30a, pMD18-T-7155和pMD18-T-7156.并电泳回收目的片段,之后用T4连接酶和回收的目的片段构建连接体系,温箱内16℃培育24 h,同时制备BL21(DE3)感受态细胞,并转化连接产物,用含有kana抗性的固体平板培养基筛选,挑阳性单菌落摇菌,并进行菌落PCR和双酶切质粒检测,送检测序,正确后命名为pET30a-7155和pET30a-7156,此为重组表达载体构建成功,用作后续蛋白表达.
1.4 目的蛋白的诱导表达
将含有已测序正确的重组表达载体(pET30a-7155和pET30a-7156)BL21菌液接种于含有kana抗性的LB液体培养基中,37℃摇床培养3 h至OD值在0.4~0.6后加入终浓度为0.6 mmol/L的IPTG于28℃下摇床培养8 h,诱导目的蛋白的表达,并用含有pET30a空载的BL21为对照.之后离心弃上清收集菌体,超声波破碎取样并加入β-巯基乙醇处理并于沸水中煮6 min,再加入5×Buffer,用SDS-PAGE电泳检测目的蛋白的表达.
2 结果
2.1 all7155和asl7156的基因克隆
用1.0%的琼脂糖凝胶电泳检测T克隆酶切后的目的条带,可见与预期大小(331 bp和258 bp)相符(见图1).由于引物设计时有酶切位点和保护碱基,故检测目的条带的位置轻微上移,送检测序pMD18-T-7155和pMD18-T-7156结果与NCBI公布的all7155和asl7156碱基序列100%吻合,证明成功克隆了目的基因.
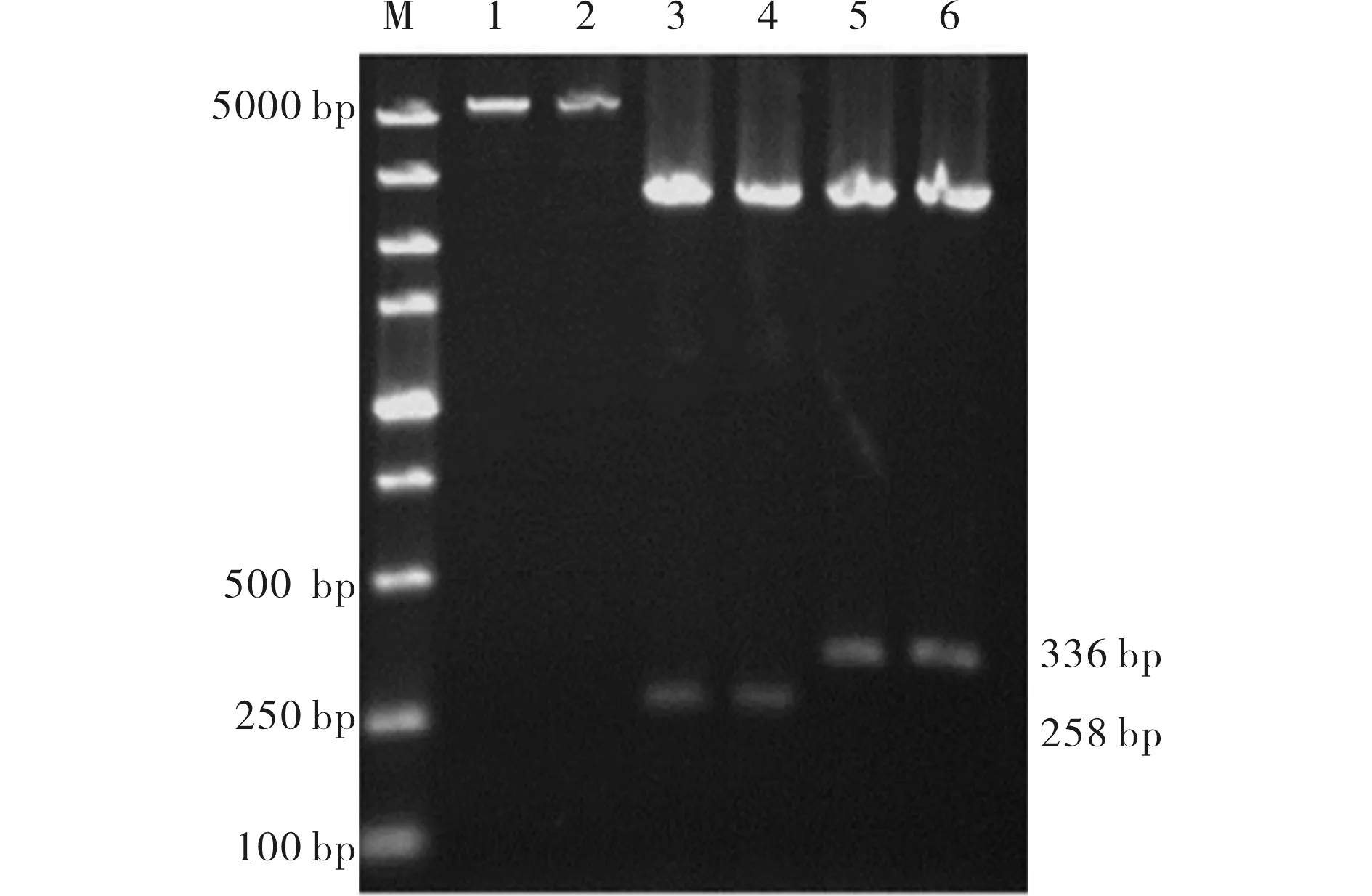
M) Marker;1,2) 为未插入目的基因的PET30a(+)载体质粒;3,4) 为插入asl7156的PMD18-T;5,6)为插入all7155的PMD18-T图1 T克隆酶切后电泳检测Fig.1 Electrophoresis identification of T clone after enzyme cutting
2.2 构建重组表达载体
挑取阳性菌落摇菌后提取质粒(见图2),根据引物设计时插入的双酶切位点BamH I/Hind III,对重组后的pET30a进行双酶切,可见与目的片段大小相符的条带(见图3),送检同批菌液测序,测序结果与NCBI中的目的基因序列100%吻合,即验证所插入片段为336 bp的all7155和258 bp的asl7156.
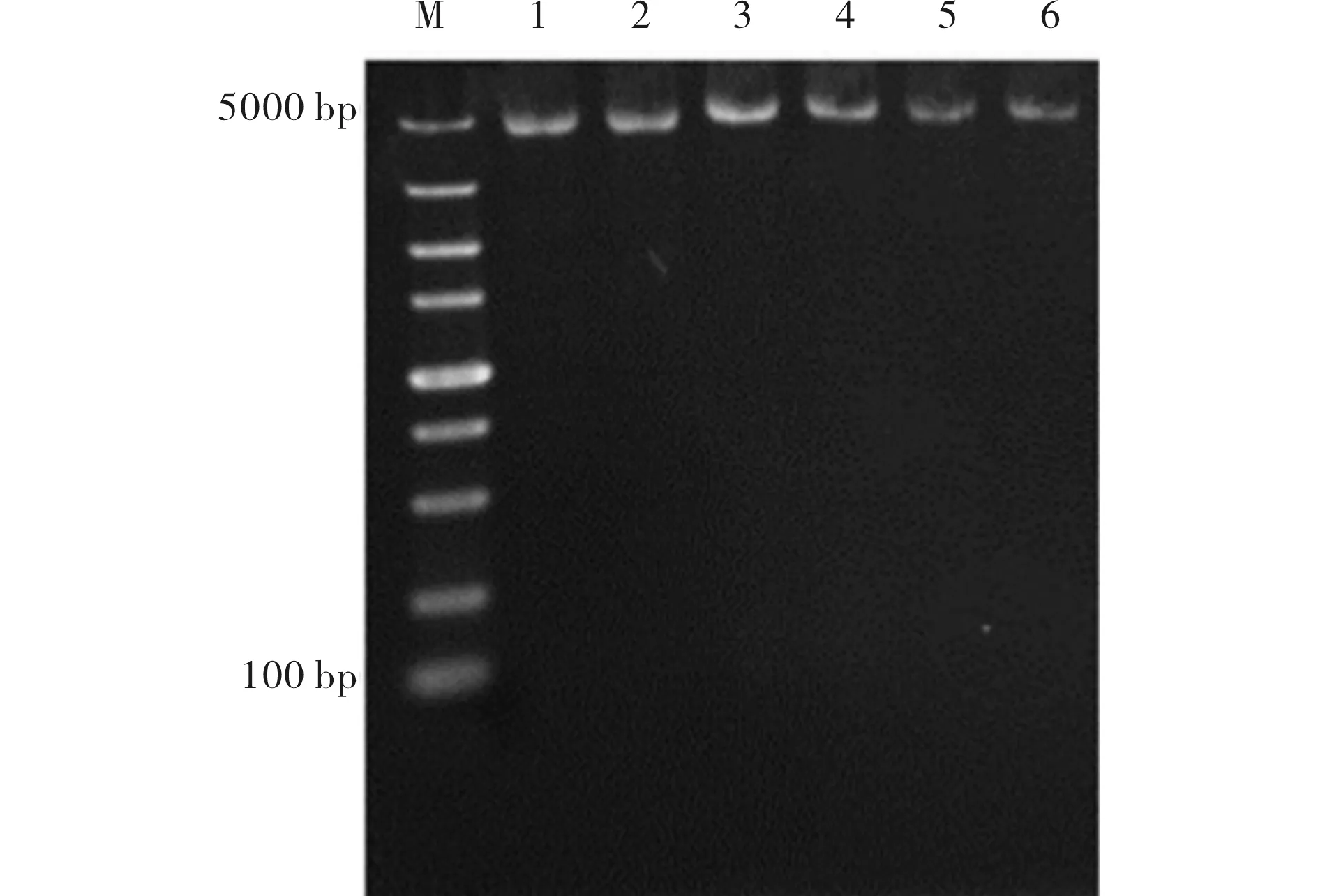
M) DNA Marker;1,2)为空载pET30a质粒;3,4)插入all7155片段的重组表达载体质粒;5,6)为插入片段为asl7156片段的重组表达载体质粒图2 电泳检测重组表达载体的质Fig.2 Electrophoresis identification of plasmid recombined with expression vector
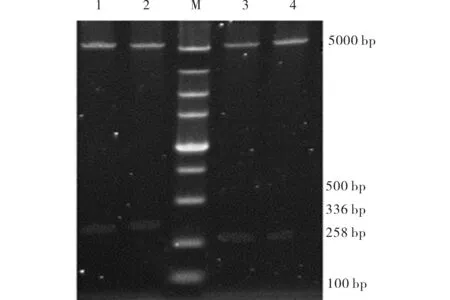
M)Marker; 1,2)为插入all7155的PET30a ;3,4)为插入asl7156的pET30a图3 重组表达载体Fig.3 Electrophoresis identification of recombinant expression vector
2.3 IPTG诱导目的蛋白的表达
将含有目的基因all7155和asl7156的重组表达载体BL21菌液在0.6 mmol/L的IPTG,28℃摇床中培养,并设置时间梯度2,4,6,8 h诱导表达,经SDS-PAGE电泳检测(见图4)可见:分别在15KD上方和20KD下方由时间不同出现一条亮度逐渐增强的条带,因预测all7155和asl7156基因的表达蛋白的分子量分别为13.2 KD和10.2 KD,又因pET30a (+) 表达载体的带有6组氨酸标签,使产生的融合蛋白的分子量增加约5KD[7],故目的条带的位置应该在15KD和20KD之间

30a IPTG) 加入了IPTG诱导的30a空载; 30a) 未加入IPTG的30a空载; M) Protein ladder图4 SDS-PAGE 检测不同时间下IPTG诱导表达all7155(图A)和asl7156(图B)Fig.4 SDS-PAGE identification of IPTG induced all7155(Fig.A) and asl7156 (Fig.B)expression at different hour
3 讨论
本文先通过NCBI中BLAST序列分析发现PCC7120α质粒上的基因all7155与大肠杆菌染色体上毒素基因ParE具有较高的同源性,而ParE在T-A系统中充当一个毒素因子,它在细胞的DNA复制进程中通过促使DNA双链解螺旋而起到毒素作用[5],且与细胞的稳定性生长密切相关.asl7156通过BLAST蛋白比对后发现其与细胞生长中诱导沉溺模式产生的基因高度同源,推测其编码蛋白起解毒作用.为此对基因构成毒素-抗毒素系统提供了理论基础,由于对ParD/E家族的性质和作用研究认识较MazE/F家族少而不全,以此对基因来进一步研究ParD/E毒素抗毒素性质具有较高的参考价值.
本文先设计带有特殊双酶切位点和保护碱基的引物,PCR克隆all7155和asl7156目的基因并纯化后连接至pMD18-T载体,阳性克隆单菌落测序后证明成功构建了目的基因的克隆载体,并通过DH5α感受态转化连接目的基因片段至pET-30a,筛选阳性单菌落测序后构建了序列正确的表达载体pET30a-7155和pET30a-7156.以BamH I/Hind III为酶切位点,主要考虑了其切割率和易连性,利用蓝白斑筛选和DH5α感受态连接转化的方法也符合技术成熟,可行可靠和重复性好的原则.
蛋白表达选择了BL21的大肠杆菌作为表达菌,尽量减低了其他不利因素的影响,通过固定一个变量,梯度设计另一变量来进行比较,在相对的低温,低转速内能提高表达量[9],IPTG在诱导表达的同时因浓度而抑制菌体生长实验确定了平衡蛋白表达和适合菌体生长的IPTG浓度.超声波破碎的结果显示:两基因表达蛋白大量存在于菌体裂解液上清中,沉淀中微量存在是由于培养过程中形成的包涵体,说明此类蛋白应为可溶性蛋白.设置时间梯度更易检测到目的蛋白在合适的IPTG浓度(0.4~0.6 mmol/L)下,随着不同培养时间表达量由少变多,并用于验证目的蛋白的表达的大小和位置.
由于选用的表达载体质粒pET-30a(+)带有His-tag,在菌体大量培养后,可利用Ni柱进行纯化,获得纯化后蛋白可研究其物理化学性质及相互作用,用于其毒素抗毒素性质的检测及定性.鉴于毒素-抗毒素系统的研究在医学领域的在兴起,如某一对毒素-抗毒素基因对在细菌生长过程对其细胞进程起关键作用,故本研究还具有一定的理论意义和医疗实用价值.
参 考 文 献
[1] Sevillano L, Díaz M, Yamaguchi Y, et al. Identification of the first functional toxin-antitoxin system in Streptomyces[J]. PloS one, 2012, 7(3): e32977.
[2] Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, et al. Bacterial programmed cell death and multicellular behavior in bacteria[J]. PLoS genetics, 2006, 2(10): e135.
[3] Oberer M, Zangger K, Gruber K, et al. The solution structure of ParD, the antidote of the ParDE toxin-antitoxin module, provides the structural basis for DNA and toxin binding[J]. Protein Sci, 2007,16(8):1676-1688.
[4] Diago-Navarro E,Hernandez-Arriaga A M,López-Villarejo J,et al.parD toxin-antitoxin system of plasmid R1-basic contributions,biotechnological pplications and relationships with closely-related toxin-antitoxin systems[J]. FEBS J,2010,277(15): 3097-117.
[5] Van Melderen L. Toxin-antitoxin systems: why so many,what for [J].Curr Opin Microbiol,2010,13(6): 781-785.
[6] Pandey D P,Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes[J]. Nucleic Acids Res,2005,33(3): 966-976.
[7] 刘 欣,陈思礼,梅 菊,等.集胞藻PCC6803基因sll0853的克隆优化表达及其结构功能预测[J].山西大学学报,2012,35(3):552-556.
[8] 季建军,邱景富,杨瑞馥. 细菌的细胞程序性死亡中毒素-抗毒素系统的研究进展[J]. 军事医学科学院院刊,2006,30(2):184-187.
[9] 任增亮,堵国成,陈 坚,等. 大肠杆菌高效表达重组蛋白策略[J].中国生物工程杂志,2007,27(9):103-109.
[10] 萨姆布鲁克J,拉塞尔D W.分子克隆实验指南精编版[M].黄培堂,王恒樑,周晓巍,等,译.3版.北京:科学出版社,2007.
