非负矩阵分解在微阵列数据分类s和聚类发现中的应用
2014-08-03任重鲁李金明
任重鲁,李金明
(南方医科大学基础医学院生物信息学系,广东 广州 510515)
1 引言
微阵列技术源于斯坦福大学的cDNA芯片和Affymetrix公司的寡核苷酸探针技术,经过十多年的发展,现在已经成为生命科学研究中不可或缺的重要手段[1],其高通量的特性成为研究人类疾病的有力工具。微阵列技术大致可以分为基因芯片、microRNA芯片、甲基化芯片以及蛋白结合芯片等,这些芯片产生了海量的待处理和分析数据。如何对微阵列数据进行准确而合理的分析,已成为有效应用微阵列技术的瓶颈问题,并已成为当前生物信息学的重要研究内容和研究方向[2]。
在人类疾病中,癌症(恶性肿瘤)因为较高的发病率和死亡率继续成为全球的负担[3]。癌症具有高度异质性,形态学相似、临床表现相似的癌症很可能需要不同的治疗方案[4],相同病理分期的癌症却有着不同的预后表现[5~6],这些都给癌症的临床治疗和术后辅助化疗带来不确定性。从分子生物学的角度去揭示癌症异质性,划分不同癌症亚型,用以辅助临床诊断、治疗癌症具有重要意义。在以往的研究中,科学家利用DNA微阵列或基因芯片技术及其后续数据分析,系统地揭示了卵巢癌[7]、前列腺癌[8]、乳腺癌[9]、胶质细胞瘤[10~11]等的不同亚型、预后相关的基因标签、复发相关的基因标签,癌症的发生发展以及转移相关[12~13]的基因和通路。主要使用的数据挖掘方法是对基因表达谱进行聚类发现和类别预测分析。通过基因筛选或特征提取获得信息基因,用之建立分类器,然后再考查信息基因的功能来对癌症亚型进行生物学的定义。
在数学上,微阵列数据一般为N×M的矩阵,N表示基因或者探针数量,M表示样本数量,往往M≪N,也就是具有典型的小样本、高维度的特点[14]。矩阵分解[15]可以表示成:
D=M+ε=AP+ε
(1)
其中,D表示原有微阵列数据,M表示通过因式分解重建的数据矩阵,A提供了在各模式(Metagenes)中基因的分布,P则表示不同模式的度量,也就是不同模式的表达谱(Metagene Expression Profiles),是原有数据同重建数据之间的误差。广义上讲,应用在微阵列数据分析中的主成分分析PCA(Principal Component Analysis)、奇异值分解SVD(Singular Value Decomposition)、独立分量分析ICA(Independent Component Analysis)、网络分量分析NCA(Network Component Analysis)、非负矩阵分解NMF(Non-negative Matrix Factorization)、贝叶斯分解BD(Bayesian Decomposition)等方法都属于矩阵分解的范畴。
本文对NMF算法做深入阐述,系统地回顾其在微阵列数据分类分析和聚类发现中的应用,NMF在算法上做出的改变和扩展,秩的确定方法,以及现有的主要分析软件。总结了各种NMF方法在经典数据集中的应用结果。最后对NMF算法的应用和结果做了相应的讨论。
2 NMF在微阵列数据分类中的解释和计算方法的改进
2.1 NMF在微阵列数据分类中的解释
1999年,Lee D D和Seung H S[16]首先将NMF方法应用在了图像的特征识别上。并于2001年给出了NMF的算法实现[17],通过多重迭代的策略使得公式(1)逐步逼近原始数据,从而建立了NMF的应用基础。其实早于Lee D D和Seung H S的工作,Paatero和Tapper就把正矩阵分解PMF(Positive Matrix Factorization)的方法应用在了对环境科学和天体物理的数据降维处理上了[18]。Kim P M和Tidor B[19]第一次将NMF方法用于大尺度基因表达数据的分析,把拥有6 316个基因300个样本的数据用50个NMF维度表示出来,同时试图注释每一个NMF维度中的基因簇并预测不同维度之间的功能联系。之后Brunet J P[20]明确定义了原始数据分解后形成的两个矩阵的生物学含义,以及秩的确定方法。将公式(1)中的字母替换成NMF习惯的表示方法,D、A、P分别替换成X、W、H,得到:
X=WH+ε
(2)
其中,X是N×M的原始微阵列数据矩阵,有N个基因、M个样本;W和H分别是N×k和k×M的非负矩阵;W中的每一列定义成一个元基因(Metagene),wij表示组成第j个元基因的原始基因i的系数;H中的每一列表示各元基因在每一个样本中的表达值,而hij表示了第i个元基因在第j个样本中的表达水平,也就是说矩阵H存储了每一个元基因的表达谱,有几个元基因那么样本就被分成了几类;k就是元基因的数量,同时也是样本分类确定的数目。那么,把微阵列数据进行非负矩阵分解之后,确定了合适的k值也就确定了样本分类数目,从而达到了给样本分类的目的。
相比于传统的矩阵分解方法,NMF方法有以下几个优点:
(1)矩阵元素非负,可直观地解释每个组成成分,比如上面提到的元基因解释方法。
(2)NMF通常会得到稀疏矩阵的结果,它把原始数据尽可能地压缩,可以很好地通过数量不多的标签基因对样本进行分类。
(3)NMF结果中的每个分量之间都不是正交的,这同SVD、PCA等方法不同,分量之间有重叠的基因或许是属于多个代谢通路或生物学过程[21];另外Kim M H和Seo H J[22]等人通过在五个基因表达数据集上比较六种矩阵分解方法(其中两种正交分解方法,四种非正交分解方法)和K-均值算法发现,非正交的矩阵分解方法在微阵列数据聚类中明显优于正交的矩阵分解方法。
2.2 标准算法和扩展
2.2.1 标准算法
Lee D D和Seung H S给出了NMF的标准算法:
步骤1选定秩k。
步骤2随机正整数初始化公式(2)中的W和H,维数分别是N×k和k×M。
步骤3迭代直到满足终止条件为止:

(3)
(4)
③标准化W的每一列。
终止条件是使得目标函数:
‖X-WH‖2=∑ij(Xij-(WH)ij)2
(5)
或者
D(X‖WH)=
(6)
达到最小化。Brunet J P对Lee D D和Seung H S的标准算法做了生物学方面的解释,并定义了元基因(Metagene)和元基因表达谱等术语,从而使得NMF方法能够真正地应用到对癌症基因组微阵列数据进行分类分析和类别预测中去。基于捕捉亚类的需要,应用在微阵列数据分类和聚类发现分析中的NMF,都应该使用公式(6)作为目标函数,因为它和相应的迭代公式(3)和(4)能更好地发现数据中的最基本结构[20]。
2.2.2 改进的算法
众所周知,由非负约束的经典算法得到的结果(H和W)本身就有稀疏性和局部代表性。然而,在分类分析中,为了得到更清晰的类边界[18]和获得几乎没有重复基因的元基因[23],在加强NMF计算结果的稀疏性方面人们做了大量工作[24~26]。他们有的对H矩阵进行稀疏化,有的对W矩阵进行稀疏化,还有的对H和W矩阵同时进行稀疏化。
(1)稀疏非负矩阵分解SNMF(Sparse Non-negative Matrix Factorization)。
增强NMF结果稀疏性的研究最早的工作是Hoyer于2002年提出的,他利用线性稀疏编码(Linear Sparse Coding)方法重建目标函数公式(5):
(7)
Liu等人[27]在借鉴了目标函数公式(7)之后,又进一步将目标函数公式(6)加入稀疏约束,改写成:
Xij+(WH)ij)+a∑i,jHij
其中,通过α来调节H中元素的稀疏性,从而把公式(3)改写成:
并且建议使用“稀疏非负矩阵分解(SNMF)”这一术语来对加入稀疏约束的NMF算法命名。Gao等[23]利用加强矩阵稀疏性的方法首先将SNMF算法应用在癌症数据的分类分析中。在三个经典的癌症样本集上证明了加入稀疏约束的NMF算法在错分率上要优于标准的NMF方法[20]。
(2)非平滑非负矩阵分解nsNMF(non-smooth Non-negative Matrix Factorization)。
Pascual-Montano等人[26]采用了不同的方法去加强NMF结果的稀疏性,他引进了一个平滑因子,这个平滑因子可以同时改变W和H的稀疏性,将公式(2)改写成:
X=WSH+ε
其中,
其中,S是一个k×k维的矩阵;I是单位矩阵,1是元素为1的维向量;0≤θ≤1,是控制平滑程度的参数,当θ=0时,模型就变成了标准NMF。整体算法流程同标准NMF相同,只需要在公式(3)中把W用WS替换;在公式(4)中把H用SH替换;在目标函数公式(6)中把WH用WSH替换。Carmona-Saez P等人[28~30]利用nsNMF来分析微阵列数据,并且开发了专门对微阵列数据进行聚类分析和分类分析的软件bioNMF。
(3)其它的改进算法及应用。
众所周知,核磁共振成像MRSI(Magnetic Resonance Spectroscopic Imaging)的数据无论从可解释性,还是识别特定组织的不确定性都给脑肿瘤的病理确诊带来了挑战。Li等人[31]利用改进的hNMF(hierarchical Non-negative Matrix Factorization)方法来分析人类脑瘤的MRSI数据,该方法能够精确地识别出三种脑肿瘤区的组织类型(正常、肿瘤和坏死)。Ortega-Martorell S等人[32]也利用改进的Convex-NMF(Convex Non-negative Matrix Factorization)方法对脑肿瘤的MRSI数据进行划分,该方法对大多数的研究样本具有高度的敏感性和特异性,能够利用有效的阈值安全地区分肿瘤和非肿瘤区域。
另外,Lee等人[33]在研究PPARs基因的毒理作用时使用Simultaneous NMF(Simultaneous Non-negative Matrix Factorization)方法对多重、多维基因芯片表达数据进行分解。该方法在四个数据集上同时进行矩阵分解,发现了新的关于PPARs基因的代谢过程和药理作用,该结果提示可以在药物发现过程中进行早期的毒性探测。
2.3 初始化方法和秩的确定
NMF这种局部最优的迭代算法,对初始化的W和H选择很敏感。随机初始化的和矩阵有时得不到全局最优的结果[34],就使得分类结果很难找到生物学含义。一般的做法是多次运行随机初始化的数据,然后保留拥有最小目标函数值的那一组分解结果。另外,也有人尝试用独立分量分析获得的矩阵中的非负元素来作为W和H的初始化。而目前应用在微阵列数据处理方面的NMF初始化一般还沿用Lee D D和Seung H S的标准算法,这就给另一种方法的引入带来了契机,这一方法同时解决了选定秩k的问题。
2003年Monti S等人[35]利用重采样方法来评估非监督聚类结果的一致聚类CC(Consensus Clustering)概念被Brunet J P用在了解决NMF因随机初始化而导致不稳定的结果上,并且定义了同型相关系数CCC(Cophenetic Correlation Coefficient)来定量地衡量聚类结果稳定性。给定一个M×M的连通矩阵C,如果样本i和j属于同一类,元素cij=1,否则cij=0。NMF算法多次随机初始化运行,就会得到连通矩阵集合{C1,C2,…,Cp},p是NMF算法运行次数,那么一致矩阵中的元素为:
相应地,同型相关系数被定义成两个距离矩阵元素之间的皮尔森相关系数:
(8)
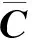
虽然Brunet J P的方法被广泛接受,还是有一些人对如何确定秩有着其它的做法。Kim P M和Tidor B[19]利用原始数据X和分解得到的WH之间的均方根误差来确定取值范围。Hutchins L N[36]特别提出在以k为横轴、残差平方和RSS(Residual Sum of Squares)为纵轴的图像里,当残差平方和出现波动的位置就是合适的k位置。Kim M H和Seo H J[22]利用间隙统计量[37]GS(Gap Statistic)来确定最优的分类数目,间隙统计量最小的时候,秩k最优。
2.4 分类结果的质量评估

Table 1 Equations for outcome assessment表1 对分类结果质量评估的几个公式
聚类有效性的评价是找到合适的度量方法来判断某一聚类划分的可接受性,也就是聚类结果要使得类内样本具有高度的相似性或尽可能地接近;而使得类间的样本具有最远的距离或尽可能地分散。以Dunn指数为例,它综合考虑了类内紧凑性和类间距离,是一个复合指数,分母表示类内最大距离,分子表示类间最小距离,那么Dunn指数越大说明聚类有效性越高。
聚类稳定性的评价是用来验证聚类算法得到结果的真实性,也就是结果在多大程度上不是因为偶然性造成的。它假设当多重样本是来自同一总体分布的抽样结果时,聚类算法将在样本上得到同总体相似的结构。以同型相关系数为例,0≤ρκ≤1,ρκ越大分类越稳定。
3 NMF算法的实现和数据分析的结果
3.1 NMF算法的实现
自从将NMF方法用于微阵列数据的分类分析和聚类发现之后,先后出现了很多个可以实现分析功能的软件。Brunet J P最早开发了基于Matlab的代码来分析微阵列数据,之后又有人写出了C++的版本[41],可在Linux操作系统下运行,但是没有被广泛使用。值得一提的是,Carmona-Saez P等人[29~30]开发了免费的图形界面bioNMF软件和基于Web网页的分析工具,该软件有三个模块组成,包括标准NMF算法、双向聚类分析、样本分类分析,其中双向聚类分析采用的是nsNMF算法。该软件操作简单,参数明确,同时结果以重排序的一致聚类图片显示,具有很强的直观性。此外,其它的免费软件还有Yamayo开发的基于GenePattern[42]的NMF分析模块,该模块有基因筛选功能和分类分析功能。Qi Q等人[43]对BRB-ArrayTools软件也添加了NMF分析模块。BRB-ArrayTools是被广泛使用的针对微阵列数据分析的集成软件包,它以Excel加载宏的形式呈现,对于不善编程的生物学家来说,用户界面友好熟悉。
另外,作者推荐Gaujoux R等人[44]编写的在R/Bioconductor平台[45]上使用的免费NMF软件包。该软件包中包含了六种NMF算法,三种初始化方法,还有三种终止条件;并且该软件包有很好的兼容性,允许使用者按照自己的需要去添加新的算法、初始化方法和终止条件,从而得到令人满意的结果。表2中列出了在微阵列数据分析中利用NMF算法的软件。
3.2 数据分析结果
在以往的研究中,使用最多的数据集是急性白血病数据集[46]、中枢神经系统肿瘤数据集和髓母细胞瘤数据集[47],三个数据集的信息在表3中给出。
Table 2 Existing implementations of the NMF algorithm for microarray analysis表2 用于微阵列数据分类分析主要的NMF算法实现
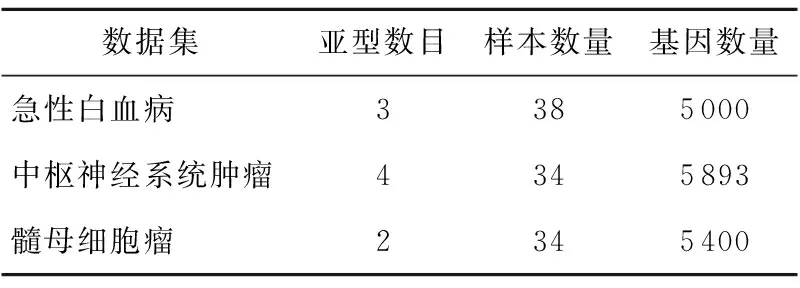
Table 3 Information about the three datasets表3 常用的三个数据集相关信息
对改进的NMF算法的评估大都通过应用如表3所示的数据集进行。本文使用急性白血病数据集来说明NMF方法(核心算法为:NMF、nsNMF)相对于其它传统方法(系统聚类、K-均值聚类)把样本归类正确率的优势。急性白血病数据集中包含三个疾病亚型,分别是AML(11例)、ALL-B-cell(19例)和ALL-T-cell(8例);共有5 000个基因的表达值在38个样本中变异最大。在R平台下使用非负矩阵分解的NMF软件包和一致聚类的ConsensusClusterPlus软件包来比较聚类结果。

Figure 1 Results of the five clustering approaches in acute leukemia dataset图1 在急性白血病数据上使用5种聚类分析方法的结果
从图1的五个分图中可以看到,图1a和图1b将38个样本明显分成三个样本簇,这三个样本簇完美地对应了急性白血病的三个亚型;而图1c中传统的系统聚类结果,无论使用何种类间度量方法都无法得到有意义的划分,说明系统聚类方法的结果无法在生物学含义上进行解释,更倾向于强行地把数据划分出层次结构;图1d和图1e则在系统聚类方法和K-均值聚类方法上使用了重采样方法,来提高分类正确率,但是图中反映出的聚类结果并不理想。表4给出了急性白血病样本集在四种方法下的分类正确率(由于系统聚类不能反映正确划分故不考虑其样本簇的正确率)。

Table 4 Performance comparisons of the four approaches表4 四种方法的分类表现
本文结果同其它研究相似,增强稀疏性的NMF算法和改变正交性的NMF算法[48]都要比最初由Lee D D和Seung H S提出的NMF算法在分类分析中的表现好。把NMF算法、经过改进的NMF算法(比如SNMF、nsNMF)同传统的矩阵分解方法(PCA、SVD等)或者层次聚类(Hierarchical Clustering)、K-均值聚类、自组织映射(SOM)等非监督聚类方法相比较[20~22],发现在对微阵列数据分类分析和聚类发现中,NMF及其经过改进的算法在分类正确率上都要好于传统的方法,并且NMF方法更容易发现数据本身具有的基本结构,而不是被诸如层次聚类这样的方法强行地把数据分出层次结构。另外,在聚类发现中NMF更多地被用来得到秩的值,也就是确定样本被分为几类和每个样本的类标签,之后再用其它的基因选择方法(PAM[49]、CLaNC[50])对有类标签的样本进行基因选择操作,从而得到重要的标签基因,并考查标签基因的生物学含义。
4 结束语
微阵列数据分类分析的目的一般来说是识别出具有生物学意义的标签基因,这些具有标签作用的基因能够对疾病的发生、发展有指示作用,进一步的探讨使得基于特定分子表达谱的个体治疗成为可能。有监督的聚类分析和非监督的分类分析是经常使用的方法。
非负矩阵分解及其扩展算法能够应用在微阵列数据分析中,使得微阵列数据分析又多了一种有力的方法。尽管非负矩阵分解有着收敛速度慢、局部最优、结果依赖初始化、算法复杂、对于数据量大的数据较耗费时间等局限性,但是它的非负约束、稀疏性约束使得计算结果更易于用生物学知识来解释。一般来说,它要比传统的聚类或分类方法更有效,因为它有助于发现微阵列数据中真正存在的层次结构。建议在使用非负矩阵分解的时候同样使用一种传统的分类方法,两种方法的结果经过对照或者综合分析之后,得到的最终结果更趋于真实。由于元基因的组成基因可以成为分类标签,非负矩阵分解还可以作为基因选择的方法;此外,尽管增加稀疏性,但是元基因之间还是会有重叠的基因出现,这些基因可能同时在多个通路或者生物学过程中,往往这种“身兼数职”的基因在要分析的问题中有更为重要的意义。
总之,非负矩阵分解是一种分析和解释具有大尺度性质的微阵列数据的新方法,现在越来越多地应用到实际问题中[10,51~53],尤其在近两年非负矩阵分解被广泛地应用到新的研究领域中。在宏基因组学研究中,Jiang X等人[54]利用NMF方法来探索海洋微生物的生物地理学方面的问题,用少数生态成分的线性组合来解释来自不同标本采集点的微生物的8 214个蛋白质家族。结论认为NMF筛选的方法要优于PCA筛选的方法,它揭示了不同标本采集点之间的功能距离同环境距离有很强的相关性,而跟地理距离相关性不大。另外,在研究复杂疾病方面,Wang H M等人[55]用NMF方法来研究复杂疾病的内在表型,将176例晚发老年痴呆症(Late-onset Alzheimer’s Disease)样本分成三个亚型并提取同每个亚型相关的易感性基因,并且给复杂疾病的病理机制的研究提供了新的方法,有助于更好地理解基因型和表型之间的关系。可见,随着更多克服非负矩阵分解缺陷的新方法不断出现,非负矩阵分解的应用及表现不会局限在微阵列的数据分析,它还可以应用在图像处理[31~32]、声音处理、文本挖掘、信息检索等领域。
[1] Russell S, Meadows L, Russell R. Microarray technology in practice[M].Xiao Hua-sheng,Zhang Chun-xiu, Wu Xue-mei, et al,translation. Beijing:Science Press,2010.(in Chinese)
[2] Huang De-shuang. Research on mining approaches for gene expression profiles data[M].Beijing:Science Press,2009.(in Chinese)
[3] Jemal A, Bray F, Center M M, et al. Global cancer statistics[J]. CA Cancer J Clin, 2011, 61(2):69-90.
[4] Valk P J M, Verhaak R G W, Beijen M A. Prognostically useful gene-expression profiles in acute myeloid leukemia[J]. The New England Journal of Medicine, 2004, 350:1617-1628.
[5] Barrier A, Boelle P-Y, Roser F, et al. Stage ii colon cancer prognosis prediction by tumor gene expression profiling[J]. Journal of Clinical Oncology, 2006, 24(29):4685-4691.
[6] Wang Y, Jatkoe T, Zhang Y, et al. Gene expression profiles and molecular markers to predict recurrence of dukes’b colon cancer[J]. Journal of Clinical Oncology, 2004, 22(9):1564-1571.
[7] The Cancer Genome Network. Integrated genomic analyses of ovarian carcinoma[J]. Nature, 2011, 474(7353):609-615.
[8] Taylor B S,Schultz N,Hieronymus H,et al.Integrative genomic profiling of human prostate cancer[J]. Cancer Cell, 2010, 18(1):11-22.
[9] Sorlie T, Perou C M, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications[J]. PNAS, 2001, 98(19):10869-10874.
[10] Li A, Walling J, Ahn S, et al. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes[J]. Cancer Research, 2009, 69(5):2091-2099.
[11] Verhaak R G, Hoadley K A, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in pdgfra, idh1, egfr, and nf1[J]. Cancer Cell, 2010, 17(1):98-110.
[12] Jorissen R N, Gibbs P, Christie M, et al. Metastasis-associated gene expression changes predict poor outcomes in patients with dukes stage b and c colorectal cancer[J]. Clinical Cancer Research, 2009, 15(24):7642-7651.
[13] Smith J J, Deane N G, Wu F, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer[J]. Gastroenterology, 2010, 138(3):958-968.
[14] Vinciotti V, Tucker A, Kellam P, et al. Robust selection of predictive genes via a simple classifier[J]. Appl Bioinformatics, 2006, 5(1):1-11.
[15] Kossenkov A V, Ochs M F. Matrix factorization methods applied in microarray data analysis[J]. Data Mining and Bioinformatics, 2010, 4(1):72-90.
[16] Lee D D, Seung H S. Learning the parts of objects by non-negative matrix factorization[J]. Nature, 1999, 401:788-791.
[17] Lee D D, Seung H S. Algorithms for non-negative matrix factorization[J]. Adv. Neural Inform Process System, 2001, 13:556-562.
[18] Devarajan K. Nonnegative matrix factorization:An analytical and interpretive tool in computational biology[J]. PLoS Computional Biology, 2008, 4(7):e1000029.
[19] Kim P M, Tidor B. Subsystem identification through dimensionality reduction of large-scale gene expression data[J]. Genome Res, 2003, 13(7):1706-1718.
[20] Brunet J P, Tamayo P, Golub T R, et al. Metagenes and molecular pattern discovery using matrix factorization[J]. PNAS, 2004, 101(12):4164-4169.
[21] Frigyesi A, Höglund M. Non-negative matrix factorization for the analysis of complex gene expression data, identification of clinically relevant tumor subtypes[J]. Cancer Informatics, 2008, 6:275-292.
[22] Kim M H, Seo H J, Joung J G, et al. Comprehensive evaluation of matrix factorization methods for the analysis of DNA microarray gene expression data[J]. BMC Bioinformatics, 2011, 12(Suppl 13):S8.
[23] Gao Y, Church G. Improving molecular cancer class discovery through sparse non-negative matrix factorization[J]. Bioinformatics, 2005, 21(21):3970-3975.
[24] Hoyer P O. Nonnegative sparse coding[C]∥Proc of Neural Networks for Signal Processing XII, 2002:557-565.
[25] Hoyer P O. Non-negative matrix factorization with sparseness constraints[J]. Journal of Machine Learning Research, 2004, 5:1457-1469.
[26] Pascual-Montano A,Carazo J M,Kochi K,et al.Nonsmooth nonnegative matrix factorization (nsnmf)[J]. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2006, 28(3):403-415.
[27] Liu W, Zheng N, Lu X. Non-negative matrix factorization for visual coding[C]∥Proc of the IEEE International Conference on Acoustics, Speech and Signal Processing, 2003:293-296.
[28] Carmona-Saez P, Pascual-Marqui R D, Tirado F, et al. Biclustering of gene expression data by non-smooth non-negative matrix factorization[J]. BMC Bioinformatics, 2006, 7:78-96.
[29] Mejia-Roa E, Carmona-Saez P, Nogales R, et al. Bionmf:A web-based tool for nonnegative matrix factorization in biology[J]. Nucleic Acids Res, 2008, 36(Web Server issue):W523-W528.
[30] Pascual-Montano A, Carmona-Saez P, Chagoyen M, et al. Bionmf:A versatile tool for non-negative matrix factorization in biology[J]. BMC Bioinformatics, 2006, 7:366-374.
[31] Li Y, Sima D M, Cauter S V, et al. Hierarchical non-negative matrix factorization (hnmf):A tissue pattern differentiation method for glioblastoma multiforme diagnosis using mrsi[J]. NMR Biomed, 2013, 26(3):307-319.
[32] Ortega-Martorell S, Lisboa P J, Vellido A, et al. Convex non-negative matrix factorization for brain tumor delimitation from mrsi data[J]. PLoS One, 2012, 7(10):e47824.
[33] Lee C M, Mudaliar M A, Haggart D, et al. Simultaneous non-negative matrix factorization for multiple large scale gene expression datasets in toxicology[J]. PLoS One, 2012, 7(12):e48238.
[34] Shi Jin-long, Luo Zhi-gang. Research on the advances of nonnegative matrix factorization and its application in bioinformatics[J]. Computer Engineering & Science, 2010, 32(8):117-123.(in Chinese)
[35] Monti S, Tamayo P, Mesirov J, et al. Consensus clustering:A resampling-based method for class discovery and visualization of gene expression microarray data[J]. Machine Learning, 2003, 52:91-118.
[36] Hutchins L N, Murphy S M, Singh P, et al. Position-dependent motif characterization using non-negative matrix factorization[J]. Bioinformatics, 2008, 24(23):2684-2690.
[37] Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic[J]. J.R.Statist, 2001, 63:411-423.
[38] Rousseeuw P. Silhouettes:A graphical aid to the interpretation and validation of cluster analysis[J]. Journal of Computational and Applied Mathematics, 1987, 20:53-65.
[39] Frades I, Matthiesen R. Overview on techniques in cluster analysis[J]. Methods in Molecular Biology, 2010, 593:81-107.
[40] Kim H, Park H. Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis[J]. Bioinformatics, 2007, 23(12):1495-1502.
[41] Wang G, Kossenkov A V, Ochs M F. Ls-nmf:A modified non-negative matrix factorization algorithm utilizing uncertainty estimates[J]. BMC Bioinformatics, 2006, 7:175-184.
[42] Reich M, Liefeld T, Gould J, et al. Genepattern 2.0[J]. Nature Genetics, 2006, 38:500-501.
[43] Qi Q, Zhao Y, Li M, et al. Non-negative matrix factorization of gene expression profiles:A plug-in for brb-arraytools[J]. Bioinformatics, 2009, 25(4):545-547.
[44] Gaujoux R, Seoighe C. A flexible r package for nonnegative matrix factorization[J]. BMC Bioinformatics, 2010, 11:367-375.
[45] Gentleman R C, Carey V J, Bates D M, et al. Bioconductor:Open software development for computational biology and bioinformatics[J]. Genome Biology, 2004, 5(10):R80.81-R80.16.
[46] Slonim D K, Tamayo P, Mesirov J P, et al. Class prediction and discovery using gene expression data[C]∥Proc of the 4th International Conference on Computational Molecualr Biology, 2000:236-272.
[47] Pomeroy S L, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression[J]. Nature, 2002, 415:436-442.
[48] Wang Y, Jia Y, Hu C, et al. Fisher non-negative matrix factorization for learning local features[C]∥Proc of Asian Conference on Computer Vision, 2004:27-30.
[49] Tibshirani R, Hastie T, Narasimhan B, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression[J]. PNAS, 2002, 99(10):6567-6572.
[50] Dabney A R. Clanc:Point-and-click software for classifying microarrays to nearest centroids[J]. Bioinformatics, 2006, 22(1):122-123.
[51] Networks C G A. Comprehensive molecular characterization of human colon and rectal cancer[J]. Nature, 2012, 487(7407):330-337.
[52] Govi S, Dognini G P, Licata G, et al. Non-negative matrix factorization to perform unsupervised clustering of genome wide DNA profiles in mature b cell lymphoid neoplasms[J]. Br J Haematol, 2010, 150(2):226-229.
[53] Inamura K, Fujiwara T, Hoshida Y, et al. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrix factorization[J]. Oncogene, 2005, 24(47):7105-7113.
[54] Jiang X, Langille M G, Neches R Y, et al. Functional biogeography of ocean microbes revealed through non-negative matrix factorization[J]. PLoS One, 2012, 7(9):e43866.
[55] Wang H M, Hsiao C L, Hsieh A R, et al. Constructing endophenotypes of complex diseases using non-negative matrix factorization and adjusted rand index[J]. PLoS One, 2012, 7(7):e40996.
附中文参考文献:
[1] 史蒂夫·拉塞尔,莉萨·梅多斯,罗斯林·拉塞尔. 生物芯片技术与实践(中文版)[M]. 肖华胜,张春秀,武雪梅,等译.北京:科学出版社,2010.
[2] 黄德双. 基因表达谱数据挖掘方法研究[M]. 北京:科学出版社,2009.
[34] 石金龙, 骆志刚. 非负矩阵算法及其在生物信息学中的应用[J]. 计算机工程与科学,2010,32(8):117-123.
