亲水/反相二维色谱法制备桔梗中的三萜皂苷
2014-08-03邢倩倩梁鑫淼
邢倩倩, 傅 青, 金 郁, 梁鑫淼
(华东理工大学药学院,上海200237)
桔梗是草本药食同源植物,其主要药理作用为消炎、止咳、祛痰、抗癌及免疫调节等。桔梗的主要化学成分为三萜皂苷、黄酮、多糖和花色苷等,其有效成分主要为齐墩果烷型三萜皂苷[1]。迄今已报道的桔梗中的三萜皂苷类成分有几十种[2],按照其母核的区别主要分为桔梗酸类、桔梗二酸类和远志酸类[3]。桔梗中以桔梗皂苷D和桔梗皂苷E含量居高,《中国药典》(2010版)和《香港中药材标准第二期》(2008版)分别以这两种皂苷作为桔梗药材质量控制的指标成分。随着分离分析技术的提高,发现桔梗中仍含有大量的未知皂苷,需要开展更深入的分离制备工作。
传统的皂苷单体制备通常选择柱层析技术。近些年来,越来越多的分离纯化技术用于桔梗皂苷单体的制备。如He等[2]使用3种不同的层析柱对总样进行前处理,然后选择反相制备色谱纯化桔梗中的三萜皂苷。高速逆流色谱作为一种快速发展的技术,亦应用于三萜皂苷的制备。Young等[4]使用高速逆流色谱分离制备了桔梗中的6种主要皂苷,且纯度均在90%以上。但是该技术更适用于简单样品或含量较高样品的制备,在涉及复杂样品及微量样品的分离制备时,通常需要与反相色谱结合使用。如In等[5]选择高速逆流色谱首先对桔梗中的微量皂苷进行粗分离,再使用反相制备色谱进行纯化,得到两种高纯度的微量皂苷。由此可见,反相色谱在三萜皂苷的纯化制备方面起着重要作用。反相色谱因其具有分离效率高、重现性好的优点,在三萜皂苷的分离分析方面应用广泛[6,7]。桔梗中所含三萜皂苷种类多,结构差异小,单使用反相色谱很难达到高效制备的需求。综上所述,对桔梗中的三萜皂苷类化合物进行分离纯化,需将反相色谱和与之具有分离正交性的方法结合,从而有效的解决分离选择性的问题。
桔梗中的三萜皂苷结构由齐墩果烷型母核连接糖链构成,同时具有亲水基团(糖)和疏水基团(母核)。由此推测,这类化合物在亲水色谱上应有较好的保留。亲水色谱模式在糖苷类化合物的分离分析上应用较多[8,9],也有学者对三萜皂苷在亲水分离模式下的分离分析进行了探讨[10]。亲水色谱与反相色谱的保留机理有很大的差异,因此这两种分离模式具有很好的正交性。Xing等[11]建立亲水/反相二维色谱模式-串联质谱检测三七中的三萜皂苷,共检测出224种皂苷。
本文在对桔梗进行水煮醇沉后,选择反相和亲水固相萃取(SPE)过程分别去除桔梗水提物中的强极性和弱极性杂质,得到三萜皂苷类组分;使用亲水制备色谱对三萜皂苷类组分进行分离,得到精细组分;然后选取代表性的精细组分,使用反相制备色谱对其进行分离纯化。本文建立的亲水/反相二维色谱方法对于复杂样品中三萜皂苷类化合物的分离纯化具有一定的借鉴意义。
1 实验部分
1.1 仪器、试剂与材料
分析型仪器:Waters Alliance高效液相色谱(美国Waters公司),包括2695四元梯度泵、2489紫外检测器、自动进样器和柱温箱。超高效液相色谱-串联四极杆质谱联用仪(ACQUITY UPLCTMSystem with a Quattro Micro MS,美国 Waters公司),采用电喷雾(ESI)离子源。
制备型仪器:Waters自动纯化系统(美国 Waters公司),包括2545二元梯度泵、2489紫外检测器、2767馏分收集器和自动进样器。
采用Masslynx 4.1操作软件对数据进行采集和处理。
样品浓缩仪器:90YY40-2GT真空旋转蒸发仪(上海上自仪转速表仪表电机有限公司);冷冻干燥机(Scientz-10N)。
试剂:色谱级乙腈购于百灵威(北京);色谱级甲醇购于禹王公司(山东);色谱级甲酸购于Acros公司(北京);工业级乙腈和甲醇购于上海禾汽化工科技有限公司;制备级乙腈购于安徽时联特种溶剂股份有限公司;流动相用水为 Milli-Q超纯水(Millipore,美国)。
材料:桔梗饮片购于雷允上药店(上海);亲水Click XIon及反相C1固相萃取填料(60~100 μm)、XAmide(150 mm×4.6 mm,5μm)、XAmide(150 mm×20 mm,5μm)(华谱新创科技有限公司,北京);Atlantis T3(150 mm×4.6 mm,5μm)、Atlantis PrepT3(100 mm×30 mm,5μm)(Waters,美国)。
1.2 实验方法
1.2.1 样品前处理
称取2 kg桔梗饮片,加入20 L水煎煮2 h后抽滤取滤液,缓慢滴加乙醇至体积分数为75%,在4℃下静置过夜。抽滤,取滤液旋蒸,除去滤液中乙醇后用反相SPE柱(20 g/60 mL,C1,60~100μL)处理。反相SPE柱先经甲醇活化,再用水平衡后上样,分别用甲醇-水(1∶9,如无特殊说明均为体积比)和甲醇洗脱,收集甲醇洗脱部分。继续使用亲水SPE柱(20 g/60 mL,Click XIon,60~100μL)处理。亲水SPE经水活化和乙腈平衡后,取反相SPE处理后的样品上样,用乙腈-水(6∶4)洗脱,收集即得桔梗中三萜皂苷类组分。
1.2.2 色谱条件
分析型亲水色谱条件:色谱柱为XAmide(150mm×4.6 mm,5μm);流动相为水(A)和乙腈(B)。梯度洗脱条件如下:0到30 min,B相从95%到60%;30到35 min,B相保持在60%。流速为1.0 mL/min;检测波长为203 nm;进样量为20μL。
亲水色谱制备过程在Waters自动纯化系统上完成,样品质量浓度为250 g/L;所用色谱柱为XAmide(150 mm×20 mm,5μm);梯度条件与分析级别相同;流速为20.0 mL/min;触发波长为203 nm。
分析型反相色谱条件:色谱柱为Atlantis T3(150 mm×4.6 mm,5μm);流动相为水(A)和乙腈(B)。梯度洗脱条件如下:0到30 min,B相保持在20%;30到35 min,B相从20%到95%。流速为1.0 mL/min;检测波长为203 nm;进样量为5μL。
反相制备过程在Waters自动纯化系统上完成,样品质量浓度为100 g/L;所用色谱柱为Atlantis T3(100 mm×30 mm,5μm);梯度条件和检测波长均与分析级别相同;流速为42.5 mL/min。
色谱-质谱分析条件:色谱柱为Atlantis T3(150 mm×4.6 mm,5μm);流动相为0.1%(v/v)甲酸水溶液(A)和0.1%(v/v)甲酸乙腈溶液(B)。梯度洗脱条件如下:0到30 min,B相保持在20%;30到35 min,B 相从 20% 到 95%;流速为 1.0 mL/min。电喷雾电离源,正离子检测模式,脱溶剂气为氮气,脱溶剂气流速为600 L/h,脱溶剂温度为380℃,离子源温度为120℃,毛细管电压为2.5 kV,锥孔电压为15 V。
2 结果与讨论
2.1 样品前处理
由于桔梗皂苷结构中多数含有5个以上的糖单元,极性相对较大,故选定水提醇沉的提取方式。醇沉过程可除去占提取液60%(质量分数)的杂质。醇沉的提取液颜色依然较深,反相色谱分析结果显示其中仍存在大量的强极性物质(见图1a)。通过反相SPE处理,甲醇-水(1∶9)洗脱,可以去除其中的强极性物质,再经甲醇洗脱得到所需要的组分。经过此过程处理得到的样品仍含有一定量的色素及少量的弱极性物质。选择亲水SPE将色素吸附在SPE柱上,在乙腈-水(6∶4)洗脱条件下,得到三萜皂苷类组分(见图1b)。连续反相、正相SPE处理后,可避免提取液中的杂质对后续亲水色谱和反相色谱分离的干扰。
2.2 三萜皂苷类组分在亲水色谱条件下的分离

图1 桔梗提取物处理(a)前、(b)后的反相色谱分析谱图Fig.1 Analytical RPLC chromatograms of(a)pre-and(b)post-pretreatment extract from Platycodon grandiflorum
三萜皂苷在亲水色谱中的保留规律与其结构上的糖个数密切相关,通常随着糖个数的增加,其保留增强。由于亲水色谱对三萜皂苷的分离度不及反相色谱,选定亲水色谱作为第一维对三萜皂苷类组分进行分离。
首先进行色谱柱和流动相两个参数的优化。基于三萜皂苷结构特性,选择酰胺固定相(XAmide)对其进行分离。亲水模式下,水和乙腈是常用的流动相[12]。研究发现,在水中加入三氟乙酸和甲酸铵,三萜皂苷的保留时间及分离度都没有明显改善,所以选定流动相为乙腈和水,分析谱图如图2所示。
进一步,将分析条件放大到制备级别。选定制备柱型号为XAmide(150 mm×20 mm),为使制备柱达到与分析柱类似的分离效果,需保证在相同的梯度条件下两种色谱柱具有相同的线速度。相应流速可由公式(1)计算:

其中,FAnal和DAnal分别代表分析型系统中洗脱液的流速和色谱柱内径,FPrep和DPrep分别代表制备型系统中洗脱液的流速和色谱柱内径。根据该公式计算,制备条件下流速为18.9 mL/min。考虑到制备型仪器泵的精确性,选定流速为20.0 mL/min。
在制备过程中进样体积较大,为了避免穿透现象需选择合适的上样溶剂。三萜皂苷在甲醇、乙醇、正丁醇等中等极性溶剂中溶解性较好,考虑到在亲水色谱中甲醇的洗脱能力比水弱,所以以甲醇为上样溶剂。其相应的制备色谱图如图3所示,与分析谱图(图2)相似,分离效果有所下降。

图2 桔梗中三萜皂苷类组分的亲水色谱分析谱图Fig.2 Analytical HILIC chromatogram of triterpenesaponins fromPlatycodon grandiflorum

图3 桔梗中三萜皂苷类组分的亲水色谱制备谱图Fig.3 Preparative HILIC chromatogram of triterpene saponins fromPlatycodon grandiflorum
桔梗皂苷类组分按照时间触发模式,每1 min收集一次,在6~25 min内共收集20个组分。连续进样,完成20.00 g桔梗皂苷类组分的亲水色谱分离,合并相同时间组分,冻干,称重。得到各组分的质量如表1所示。
通过第一维的亲水色谱制备,大大降低了样品的复杂性。亲水色谱制备中,总上样量为20.00 g,20个组分的质量之和为15.64 g,从洗针液中回收样品1.92 g,考虑到制备过程中前5 min未收集部分,该制备方法的回收率较高。
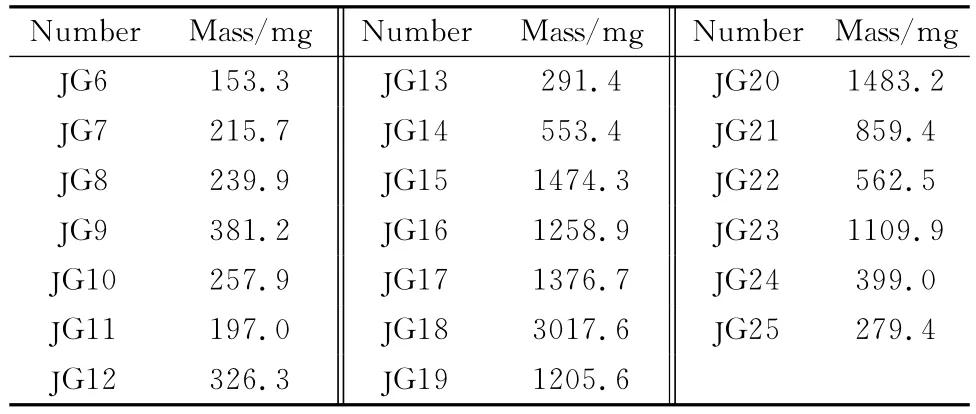
表1 亲水色谱制备得到的各个组分的质量Table 1 Mass of the preparative HILIC fractions fromPlatycodon grandiflorum
2.3 JG23组分中单体化合物的反相制备及表征
JG23为亲水色谱上收集的第18个组分,保留时间为23~24 min,质量为1.11 g。该组分在反相Atlantis T3色谱柱上分析获得的色谱图如图4所示。JG23组分在亲水色谱模式下为一个色谱峰,在反相模式下为分离度较高的两个色谱峰。由于其在两种模式下的分离呈现较好的正交性,故作为代表性组分进行制备过程示范。第二维反相色谱制备过程使用Atlantis T3 Prep色谱柱(100 mm×30 mm,5μm),根据公式(1)计算得到流速为42.5mL/min。相应制备色谱图如图4b所示,制备色谱图与分析色谱图没有明显差别。
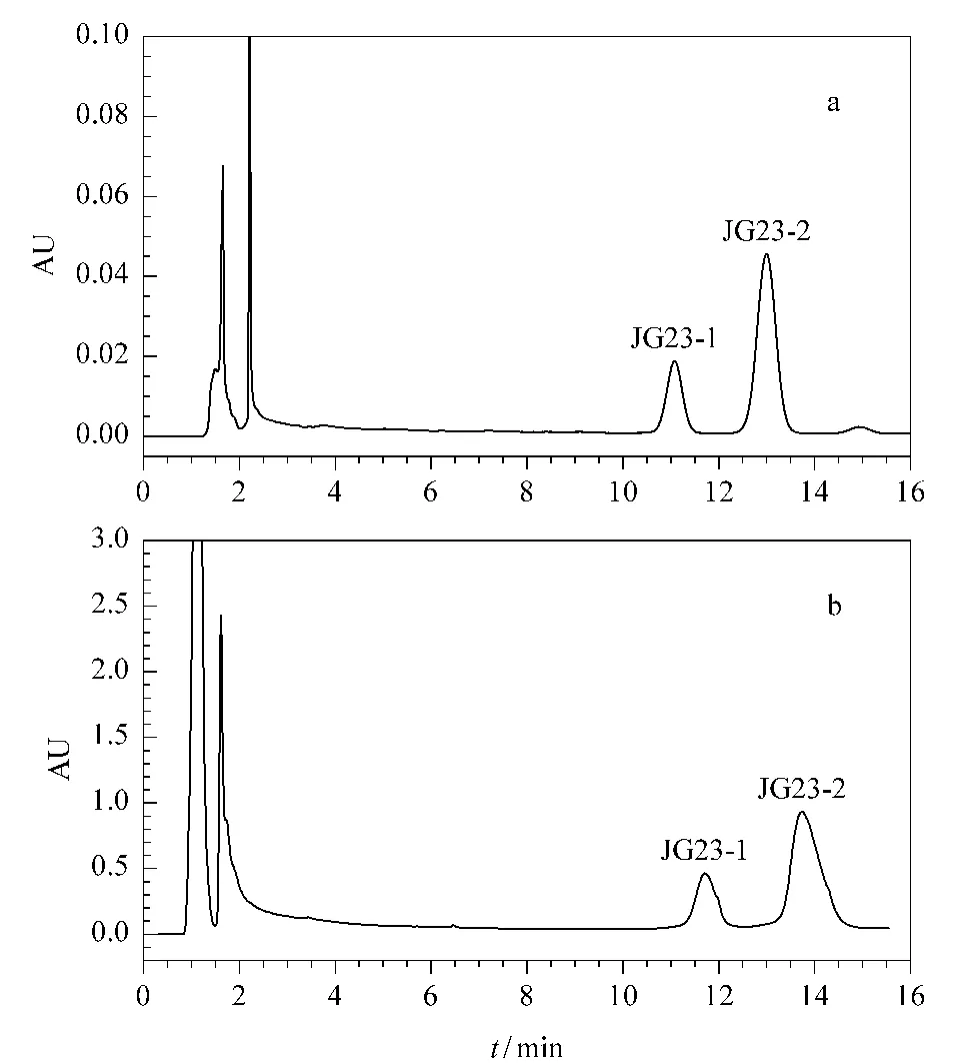
图4 优化条件下JG23组分在(a)分析和(b)制备水平的反相液相色谱图Fig.4 Analytical and preparative RPLC chromatograms of JG23 under the optimum conditions
以紫外触发的方式进行馏分收集,收集的馏分经过处理,得到单体化合物JG23-1和JG23-2,质量分别为26.2 mg和135.9 mg,色谱峰纯度分别为91.42%和98.43%。以质谱和核磁共振对其进行分析。质谱结果如图5所示,根据正离子模式下形成的[M+H]+峰推断其相对分子质量分别为1 416和1 548。通过质谱解析,确定JG23-1结构上糖单元信息为3个六碳糖、1个脱氧六碳糖和2个五碳糖;JG23-2结构上糖单元信息为3个六碳糖、1个脱氧六碳糖和3个五碳糖。结合核磁数据(未给出),确定两个化合物分别为deapi-platycoside E和platycoside E(桔梗皂苷E)[13](见图6)。

图 5 JG23-1和JG23-2的质谱图Fig.5 Mass spectra of JG23-1 and JG23-2

图 6 JG23-1和JG23-2的化学结构Fig.6 Chemical structures of JG23-1 and JG23-2
3 结论
本文以桔梗药材为研究对象,结合色谱填料建立前处理方法,除去其中的非皂苷杂质,得到三萜皂苷类组分。构建了亲水/反相二维色谱法对三萜皂苷类组分进行分离纯化,成功得到两个单体化合物,经分析其纯度均达到90%以上。使用质谱和核磁共振对其定性,确定这两种化合物分别为deapiplatycoside E和platycoside E。本文所用方法步骤少、简便,对于复杂样品中皂苷的分离纯化具有较好的实际应用价值。
[1] Zhao X L.China Condiment(赵秀玲.中国调味品),2012,37(2):5
[2] He Z D,Qiao C F,Han Q B,et al.Tetrahedron,2005,61:2211
[3] He M L,Cheng X W,Chen J K,et al.Traditional Chinese Medicine New Drug and Clinical Pharmacology(何美莲,程小卫,陈家宽,等.中药新药与临床药理),2005,16(6):457
[4] Young W H,Yeong S K.Phytochem Anal,2009,20:207
[5] In J H,Minseok K,Yun C N,et al.J Sep Sci,2011,34:2559
[6] Zhu F X,Jia X B,Cai Y,et al.Acta Chromatographica,2013,25(1):147
[7] Sun B S,Gu L J,Fang Z M,et al.J Pharm Biomed Anal,2009,50:15
[8] Zhang J,Wang L L,Shan L G,et al.Chinese Journal of Chro-matography(张静,王玲玲,单联国,等.色谱),2012,30(8):804
[9] Shi Y M,Yang H,Li Y Y,et al.Food Research and Development(史一鸣,杨虹,李燕艳,等.食品研究与开发),2012,33(12):134
[10] Noel S Q,Nerissa L D,Azamjon B S,et al.Anal Bioanal Chem,2007,389:1477
[11] Xing Q Q,Liang T,Shen G B,et al.Analyst,2012,137:2239
[12] Boguslaw B,Sylwia N.Anal Bioanal Chem,2012,402:231
[13] Li W,Zhang W,Xiang L,et al.Molecules,2010,15:8702
