胡枝子属部分植物的ITS序列分析
2014-07-20赵月梅高明杰
赵月梅,高明杰
(商洛学院 生物医药与食品工程学院,陕西商洛726000)
胡枝子属部分植物的ITS序列分析
赵月梅,高明杰
(商洛学院 生物医药与食品工程学院,陕西商洛726000)
利用ITS序列分析技术对胡枝子属植物进行了亲缘关系分析,探讨其遗传多样性及其亲缘关系,试图为胡枝子属植物资源开发利用提供依据。将从GenBank检索获得的24条胡枝子属植物(19个种)的ITS序列做系统发育分析,Clustal 2.0软件进行序列比对,Mega4.0计算序列核苷酸比例及遗传距离,并构建邻接树(Neighbor-joining tree,NJ Tree)。胡枝子的19种植物ITS序列全长为622 bp,变异位点占总序列的16.6%;K-2-P遗传距离为0.001-0.085;鉴定成功率为57.9%,19种胡枝子可以聚为3支,与形态学分类结果并不完全一致。
胡枝子属;ITS序列;系统发育
胡枝子属(Lespedeza) 为蔷薇目豆科(Leguminosae),目前约60个种,其野生种质资源丰富,分布于世界各地。该属植物用途十分广泛:常用于水土保持和土壤改良等[1],具有抗炎、抗过敏、镇痛等药理活性[2-3]。随着分子生物学技术飞速发展,DNA分子标记技术的应用为植物的遗传结构及物种鉴定等的研究提供了高效而可靠的办法。高等植物的rDNA(ribosomeDNA)是由核糖体基因及与之相邻的间隔区组成的高度重复串联序列,其基因组序列从5’到3’依次为:外部转录间隔区(external transcribed spacer,ETS)、18S rDNA、 内 部转录间隔区1(internal transcribed spacer,ITS1)、5.8S rDNA、内部转录间隔区2(ITS2)、26S rDNA和非转录间隔区(non transcribed,NTS)。其中18S、5.8S和26S的基因组序列由于是编码区,选择压力大,在生物种间变化小,而内转录间隔区ITS1和ITS2作为非编码区,承受的选择压力较小,序列变异较大,可以提供大量的遗传学信息[4]。因此在近缘种及易混淆的中草药中得到了广泛的应用[5-8]。
系统分类上将胡枝子属植物分为两个大组:大胡枝子组(Macrolespedeza)和胡枝子组(Lespedeza)[9]。多名学者针对其分类和遗传特性利用各种分子生物学技术标记进行了研究[10-12],结果表明胡枝子属植物的种间多态性十分丰富。胡枝子属植物中存在大量的经济植物及药用植物,但是由于表型变异大,导致该属植物分类上的标准比较模糊。本研究中拟对部分胡枝子属植物进行ITS序列测定,旨在从分子生物学水平上重建胡枝子属植物的ITS系统发育树,探讨其遗传多样性及其亲缘关系,为胡枝子属植物种植资源开发利用和遗传多样性的保护提供参考依据。
1 材料与方法
1.1 材料
本研究所采用的宽叶胡枝子等19种胡枝子的24条ITS序列均从genebank下载,用ClustalX 2.0软件[13]对所有序列进行对位排列,经手工校正获得目的片段。经比对剪切后共获得24条序列,具体信息见表1。

表1 GenBank下载序列信息
1.2 数据处理
1.2.1 序列比对
将下载好的24条ITS序列导入ClustalX 2.0软件,进行多序列对比,仔细检查比对结果是否合理,去除序列中模糊的部分和前后长度不一致的序列,将所有序列剪切至相同长度,其中空位(gap)作丢失(miss)处理,找出所有序列中的变异位点。
1.2.2 计算种内、种间的遗传距离与进化树
用MEGA4.0软 件[14]基 于K2-P (Kimura2-parameter)双参数模型,采用NJ法(1000次重复bootstrap检验各分支的支持率),将比对过的序列构建系统发育树以便直观鉴定物种。
1.2.3 柱形图的构建
判断某段序列是否符合条形码的标准是确定其种内距离和种间距离之间的gap,本文用Meier等[15]的TaxonDNA软件分别计算出每个物种的种内距离和种间距离,并结合一般的统计软件构建出能够体现种间、种内的遗传距离的分布频度的柱形图。
2 结果与分析
2.1 ITS序列碱基组成分析
将得到的ITS序列进行剪切,序列比对及碱基替换分析(空位Gap作缺失处理),获得约622 bp长度的序列,对齐比对后,发现该序列存在较多的变异位点及缺失位点。其中序列中T、G、A、C的平均含量分别为19.6%、31.5%、17.4%、31.5%,G+C含量为63%,变异位点有103个,占总序列的16.6%,有50个碱基的插入和缺失,整个ITS区的变异位点丰富,可以较好地区分大部分不同种质来源的胡枝子属植物。
2.2 ITS序列遗传距离分析
胡枝子属种间K2P距离分析见表2。

表2 胡枝子属种间K2P距离
由表2可见,不同种质来源胡枝子的K2P遗传距离范围为0.001-0.085。其中,绿叶胡枝子和同裂胡枝子遗传距离最小,为0.001,表明二者亲缘关系十分相近,其次是绿叶胡枝子和大叶胡枝子,二者之间遗传距离为0.002,同裂胡枝子和短梗胡枝子之间以及长叶胡枝子与美丽胡枝子之间距离均为0.004,表明这几对物种之间亲缘关系较近,进化时间较短。另外,截叶铁扫帚与头状胡枝子之间距离最大,为0.085,表明两者遗传关系最远,宽叶胡枝子和头状胡枝子之间遗传距离次之,为0.080,表明这两个物种之间进化相对独立。
由图1可知,ITS序列在胡枝子属植物中的种内种间遗传距离分布比较符合DNA条形码的筛选标准,即种内距离集中在0-1.0%,在这个距离区间无种间距离分布;种间距离多集中在3.0%之后,表明种间距离足够大。值得注意的是,在1.0%-3.0%存在二者重叠,所以还需要系统发生树的证据进一步验证ITS序列是否符合DNA条形码的标准。
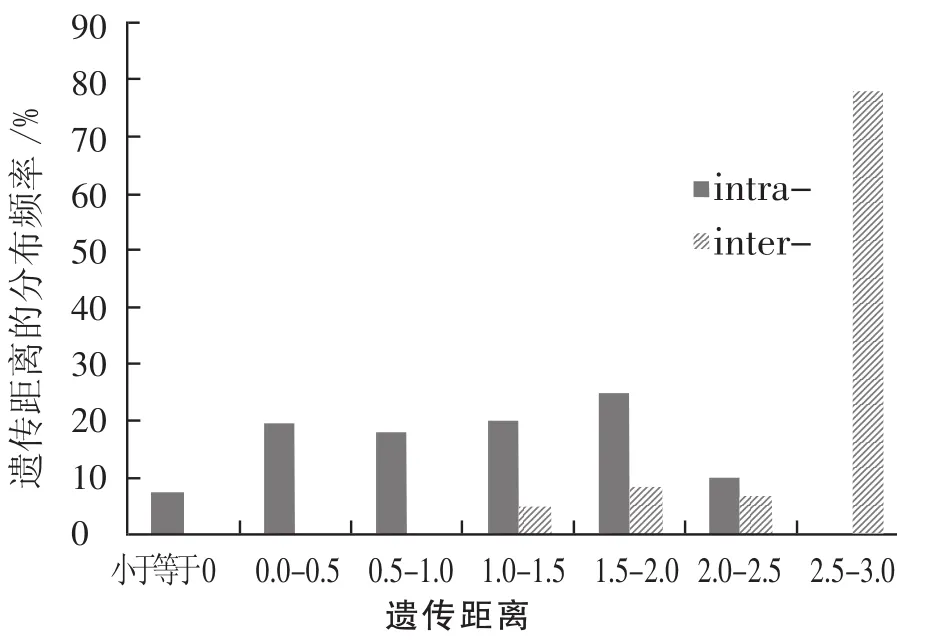
图1 胡枝子属植物种内、种间的遗传距离分布频度
2.3 系统发育分析
从GenBank下载的胡枝子属的19种胡枝子24条ITS序列做系统发育分析,在用Clustal2.0软件做多重对位排列后,去除所有序列5’端和3’端的非对位排列区,最后的对位序列共包括了空位在内的622个位置用于进化树构建。基于Mega4.0软件包的Kimura 2-paramete模式构建的系统进化树见图2(bootStrap 1000次重复,枝上数值仅显示自展支持率>50%)。
结果显示,24条胡枝子属的ITS序列分别聚为独立的3支。其中,3条兴安胡枝子序列聚在一起形成一独立分支,与尖叶铁扫帚(第二支)形成并系;第三支中的铁马鞭,截叶铁扫帚,长叶胡枝子三个种聚为一支,亲缘关系较近;此外,该支中矮生胡枝子,头状胡枝子,绒毛胡枝子,多花胡枝子,细梗胡枝子,中华胡枝子也均能独立鉴定到种。第三支的剩余物种,即美丽胡枝子,同裂胡枝子,大叶胡枝子,宽叶胡枝子,短梗胡枝子,中华垂花胡枝子,展枝胡枝子,绿叶胡枝子则形成了梳子结构,无法完全区分,美丽胡枝子的4条序列之间同源性不高,没有完全聚在一起,而是与其他几种混在一起。结果表明,ITS序列在胡枝子属的这19种植物中,有8种植物是不能用系统发育树不能完全鉴别的,剩余11种则可以鉴别到种。
3 结论与讨论
据《中国植物志》记载,分布于中国的胡枝子属植物共有26种,广布于东北到长江流域至西南地区,以及台湾山地的林缘、林迹地。在20世纪初,国外已经广泛开展了胡枝子属植物的引种和栽培技术研究,并取得了一系列的成果。我国对胡枝子的重视开始于20世纪80-90年代,主要集中在生物学特性、饲用价值和栽培技术等应用性研究上,对生物遗传学、群体遗传学等理论基础研究工作才刚刚起步。ITS区由于序列短、两端连接高度保守区、拷贝数多、长度保守、一致进化、进化速率较快等,适用于被子植物科内,尤其是近缘属间及种间甚至居群间关系的研究。在本研究中,ITS序列存在大量的种内种间变异和插入确实序列,并能够从19个胡枝子属物种中鉴别出11种,鉴定成功率为57.9%。
传统的形态分类根据有无闭锁花而将胡枝子属植物分为两组:无闭锁花的为大胡枝子组,包括美丽胡枝子、展枝胡枝子、绿叶胡枝子、宽叶胡枝子等,有闭锁花的胡枝子组包括细梗胡枝子、矮生胡枝子、尖叶胡枝子、绒毛胡枝子等。由图2可以看出,24条胡枝子序列没有完全按照传统的分类的标准聚为两组,而是聚成了三支。这一结果与传统分类上的结果冲突,原因可能为ITS序列作为核糖体基因的一部分,只能反映该序列的进化进程,并不能代表整个物种的进化历程,所以在筛选物种的DNA条形码时应尽量选择多序列联合构建邻接树,增加叶绿体和其他核糖体基因的数据,以增加鉴定上的准确性。本研究发现4条美丽胡枝子序列鉴定比较模糊,与其他几种如同裂胡枝子,大叶胡枝子等聚在了一起,这可能是因为这4条序列分布的地理距离较远,甚至出现了地理隔离等导致了遗传距离不断变大。
本文采用了ITS序列鉴别胡枝子属部分植物,以确定该序列能否作为该属植物的合适的DNA条形码,结果表明,该序列在胡枝子属植物的能达到57.9%的鉴定成功率,遗传距离在种内和种间的重叠较小,适合作为该属植物的备选DNA条形码,但是由于其成功率并没有达到100%,使用时需要与更多的片段(如matK,rbcL等)联合鉴定。

图2 基于K2P模型的ITS2序列的NJ系统发生树
[1]马彦军,曹致中,李 毅.胡枝子属植物研究进展[J].草地学报,2010,27(10):128-134.
[2]马 翼,邓虹珠,何 兰,等.甘肃胡枝子属药用植物资源[J].中国野生植物资源,2003(6):22-24.
[3]陈乃东,王春景,周守标.安徽胡枝子属植物的开发研究[J].中国林副特产,2006(1):39-41.
[4]于华会,杨志玲,杨 旭,等.药用植物种植资源ITS序列研究进展[J].中草药,2010,41(3):491-496.
[5]刘依丽,冯尚国,何仁锋,等.基于ITS2条形码对铁皮石斛及其混伪品分子鉴定的初步研究[J].杭州师范大学学报:自然科学版,2014,13(1):36-41.
[6]刘艳玲,徐立铭,倪学明,等.基于ITS序列探讨睡莲属植物的系统发育[J].武汉大学学报:理学版,2005,51(2):258-262.
[7]彭雪梅,张卫明,王蔓丽,等.罗布麻及其易混品的DNA分子鉴定[J].植物研究,2007,27(3):302.
[8]丁 平,方 琴.巴戟天与常见混伪品的rDNA ITS序列分析及其分子鉴定[J].中草药,2005,36(6):908.
[9]李翼云.中国植物志:41卷[M].北京:中国科技出版社, 1995.
[10]张吉宇,袁庆华,王彦荣,等.胡枝子属植物野生居群遗传多样性RAPD分析[J].草地学报,2006,14(3):214-218.
[11]王秀荣,赵 杨,骈瑞琪,等.胡枝子属植物ITS序列研究与系统发育分析[J].西北林学院学报,2008,23(5):70-73.
[12]赵 杨,陈晓阳,王秀荣,等.9种胡枝子亲缘关系的ISSR分析[J].吉林林业科技,2006,35(2):1-4.
[13]Larkin MA,Blackshields G,Brown N P,et al.Clustal Wand clustal X version 2.0[J].Bioinformatics,2007,23(21):2947-2948.
[14]Tamura K,Dudley J,Nei M,et al.MEGA4:Molecular Evolutionary Genetics Analysis(MEGA) software version 4.0[J].Molecular Biology and Evolution,2007,24:1596-1599.
[15]Meier R,Kwong S,Vaidya G,et al.DNA barcoding and taxonomy in Diptera:a tale of high intraspecific variability and low identification success transactions[J].Systematic Biology,2006.55:715-728.
(责任编辑:李堆淑)
Analysis of ITS Sequences in Lespedeza
ZHAO Yue-mei,GAO Ming-jie
(College of Biopharmaceutical and Food Engineering,Shangluo University,Shangluo 726000,Shaanxi)
Phylogenetic analysis of ITS in Lespedeza can explore the genetic diversity and phylogenetic relationship of this genus and provide the basis for plant resource exploitation in Lespedeza.24 ITS sequences(belong to 19 species)from GenBank Accession are obtained to analysize phylogenetic relationship,Clustal 2.0 and Mega4.0 softwares were used to caculate genetic distance and build the adjacent tree(Neighbor-joining tree,NJ Tree).The length of ITS sequences was 622bp,variation sites was 16.6%of the total sequence;K-2-P genetic distance was 0.001-0.085;identification success rate was 57.9%,19 species can be clustered into three branch,which was not entirely consistent with the morphological classification.
Lespedeza;ITS sequence;phylogeny
S541.5
:A
:1674-0033(2014)06-0068-05
10.13440/j.slxy.1674-0033.2014.06.020
2014-09-21
赵月梅,女,辽宁锦州人,博士研究生,讲师
