非绝热电子—离子耦合动力学进展
2014-07-07万世正高聪章张丰收
万世正,王 菁,高聪章,张丰收
1.射线束技术与材料改性教育部重点实验室,北京师范大学核科学与技术学院,北京100875
2.北京市辐射中心,北京100875
3.兰州重离子加速器国家实验室原子核理论研究中心,甘肃兰州730000
非绝热电子—离子耦合动力学进展
万世正1,2,王 菁1,2,高聪章1,2,张丰收1,2,3∗
1.射线束技术与材料改性教育部重点实验室,北京师范大学核科学与技术学院,北京100875
2.北京市辐射中心,北京100875
3.兰州重离子加速器国家实验室原子核理论研究中心,甘肃兰州730000
.然 Born-Oppenheimer近似的运用取得了巨大的成功,但是在非绝热跃迁过程中Born-Oppenheimer近似不再成立,所以需要重点描述电子与核之间的耦合运动。在光物理、光化学、强场物理以及离子碰撞中,非绝热过程极为常见。电子激发是非绝热过程的核心,这些过程与基态和激发态均有密切关系。原则上全量子力学方法可以准确处理非绝热电子-离子耦合,但是目前只局限于两三个原子构成的小体系。严格求解量子力学所需的计算量随着体系的增大而呈指数级增加,因此精确的量子力学处理多原子体系的非绝热过程是非常困难的。近几十年来,国内外多个小组发展了许多不同的方法来研究非绝热过程。本文重点介绍了非绝热混合量子-经典方法,即只对原子核部分的量子力学作经典极限,保留电子的量子效应。常用的混合量子-经典方法包括了Ehrenfest方法、面跳跃方法、杂化方法(即结合了Ehrenfest和面跳跃优点的方法)、基于Wigner转换的刘维尔方法。但是当原子核的量子效应显著时,非绝热动力学必须通过其它更严格的方法处理,比如从头计算多次产生方法。本文希望向刚接触非绝热动力学的同行简洁明了地阐述以上各种方法的基本思想、数值实现过程、优缺点和最新研究进展。
非绝热耦合;混合量子-经典方法;分子动力学;激发态
目录
I.引言 03
II.非绝热分子动力学中的基本概念 04
III.非绝热分子动力学 05
A.Ehrenfest方法 05
B.面跳跃方法 06
C.连续面交换方法 08
D.混合量子-经典刘维尔分子动力学 10
E.从头计算多次产生法 12
IV.Ehrenfest电子-离子关联动力学的应用 15
A.乙烯在强激光场中的响应 15
B.质子与甲醛分子碰撞 16
C.质子与甲酸分子碰撞 18
D.质子与甲烷分子碰撞 19
E.质子与乙炔、乙烯和乙烷分子碰撞 20
V.结论与展望 22
致谢 22
22
I.引言
绝热近似,又称为玻恩-奥本海默近似 (Born-Oppenheimer approximation:BOA),由玻恩和奥本海默在1927年共同提出[1]。由于电子与原子核之间质量悬殊较大,质子的质量是电子质量的1836倍,因此可以近似地将电子的运动和原子核的运动分离。绝热近似的基本思想是:当研究原子核运动时,近似地认为电子保持它的量子状态不变,也就是说电子在指定的单一量子态上运动,并且在各个能级上的概率分布恒定不变,从而研究体系在单一势能面上的运动规律。绝热近似成功地减少了求解薛定谔方程带来的巨大计算量,在量子力学研究分子光谱学和分子反应动力学中起到了至关重要的作用,同时促成了电子结构理论和分子动力学的分离。电子结构理论和分子动力学作为独立的研究方向发展取得了卓越的进展,并促进了其它相关领域的发展。
随着实验技术的发展,为观察非绝热现象提供了有效的手段;与此同时,计算机的高速发展使得人们在理论上发展了各种可能的动力学方法去探讨非绝热效应在物理和化学反应过程中的作用和意义。研究热点慢慢转向探索电子-原子核之间的非绝热耦合动力学。
虽然绝热近似已取得巨大的成功,但是根据单一的势能面推导出的动力学行为在许多情况下是不正确的,例如:在描述光解离[2]、光异构化[3]、无辐射跃迁[4,5]以及大量的高能或低能碰撞实验[6~9]等现象中。入射弹的强烈碰撞不仅会引起电荷转移现象,还会引发强激发过程,如电子逃逸和靶的库仑爆炸等现象。此外随着激光技术的发展,激光与靶分子的相互作用导致的光学响应[10]、多光子电离[11]、团簇的库仑爆炸[12]、高次谐波的产生[13,14]等现象。在研究这些光物理和光化学现象时,涉及了锥形交叉[15,16],两个电子态恰好退化,打破了绝热近似这个基本假设。因为体系中的电子不再局限于一个量子态,而是在多个量子态下运动演化,所以必须考虑电子结构与原子核运动之间的相互作用。即使是孤立的分子体系,它的预解离和内部转移现象[17,18]也涉及了多个电子态。对于大部分开壳层双原子分子,随着原子核间距离的增加,第二个电子态或更多的电子态将会产生,通常导致初始的势能面不能与产物的最低电子态一一对应[19,20],绝热近似不能适用。例如,中国科学院大连化学物理所观测了F+D2反应,发现低碰撞能激发态氟原子的反应性比基态氟高很多,这表明了绝热近似的图像对于F+D2反应是不适用的[21]。
量子力学的诞生形成了两大不同的理论框架(非线性的经典轨迹vs线性算符的期望值),从量子到经典的过渡一直是人们所关心的问题。精确求解薛定谔方程的计算量随着变量自由度数量增加而呈指数级增大,而经典力学的计算量只与自由度数量呈线性关系,这为用经典的方法描述量子现象提供了充分的理由。为了把量子的自由度融入经典的表示,许多混合量子-经典(mixed quantum-classical)的模型被提出。最早的一位践行者是Mott[22],他研究了碰撞反应中原子的激发。
本文重点关注能处理非绝热耦合的混合量子-经典方法。首先是Ehrenfest方法,该方法利用Ehrenfest定理[23]通过量子自由度的平均势计算作用于经典轨迹的有效力。其次,Tully等人提出了一种新方法:面跳跃法,经典轨迹一直在单一的绝热势能面上演化,直到满足特定的“跳跃条件”才跳到另一个绝热势能面上。第三个方法是“混合”方法,它结合了Ehrenfest方法和面跳跃方法各自的优点,这里只介绍Hack等人的连续面交换法。第四种是量子-经典刘维尔方法,它包含了密度算符的刘维尔方程和重粒子的经典Wigner极限。前面四种方法都是常用的研究电子-离子之间非绝热耦合的混合量子-经典方法。第五种方法是从从头计算分子动力学出发,引出能兼顾非绝热效应和原子核量子效应的方法:从头计算多次产生法,该方法打破了在量子动力学中作经典近似的框架,比前面的四种方法更加严格,计算量也相对较大。
文章分为五部分:第二部分推导了在混合量子-经典方法中体系的哈密顿量,并介绍了非绝热耦合的定性参考量:梅西参数。第三部分别重点阐述了以上五种非绝热方法。第四部分简单介绍了利用含时密度泛函理论与分子动力学发展的Ehrenfest电子-离子关联动力学模型,以及采用该模型研究质子与甲醛等分子碰撞的结果。最后对非绝热方法的发展进行了展望。
II.非绝热分子动力学中的基本概念
混合量子-经典方法结合了量子描述和经典力学处理,此方法在国内外都有大量的研究工作。国外如NikitinE E,Tully J C,Drukker K等人的研究本工作[24~27]:对原子核部分的量子动力学进行经典极限近似,用经典力学来描述原子核的运动; 在国内中国科学院大连化学物理研究所的韩克利组,是最早在理论上观察到了F+D2反应体系低碰撞能非绝热效应的[28,29]。其次,在本文所关注的量子经典混合方法的研究领域,该研究组也开发了可以适用于研究分子体系包括大分子体系的动力学计算程序[30~32]。
用量子力学来描述电子的运动从而保留电子的量子效应。原子核的运动轨迹R(t)和确定电子运动的含时总波函数Ψ(r;t)满足含时电子薛定谔方程:
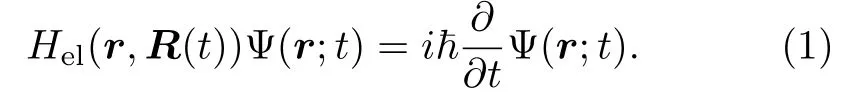
电子哈密顿量Hel包括了电子的动能,原子核-原子核、电子-原子核、电子-电子之间的势能:

其中,总波函数Ψ(r;t)是绝热本征函数Ψk(r,R)的线性组合:
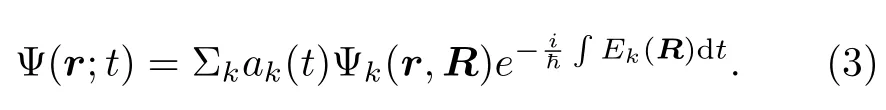
在某一确定的时间t和原子核坐标R下,Ψk(r,R)又是下面含时薛定谔方程的解:


其中Ckl包含了非绝热耦合成分,
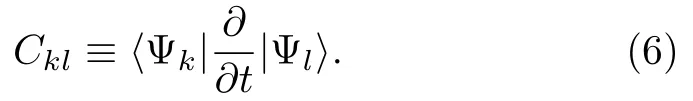
根据链式法则,Ckl可改写为:
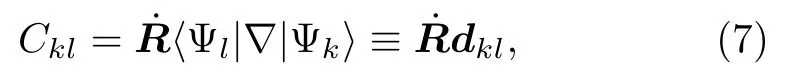
dkl是非绝热耦合矢量。 展开系数ak(t)的模平方|ak(t)|2为t时刻体系处于k绝热态的几率。
现在用最简单的二态模型来定性地讨论绝热近似的适用条件,图1给出了NaCl分子的避免交叉势能面[33,34]。一般情况下,当Na和Cl原子核间距离R较小时,NaCl分子呈现离子态:Na++Cl-;当R增大时,分子变为共价态:Na+Cl。这里构建了两个避免交叉的绝热态:Na++Cl-前段和Na+Cl后端构成了绝热基态,所对应的波函数为Ψ1;Na+Cl前段和Na++Cl-后端构成了绝热激发态,它对应的波函数为 Ψ2。波函数Ψ1和Ψ2随键长l(特征长度)和原子核构型的变化而变化,特别是在避免交叉的附近(该区域称为非渐近区域或强耦合区域),波函数在两个绝热态之间变化剧烈。当原子间间距很大时,即进入了渐进区域,非绝热效应减弱,波函数几乎保持不变。
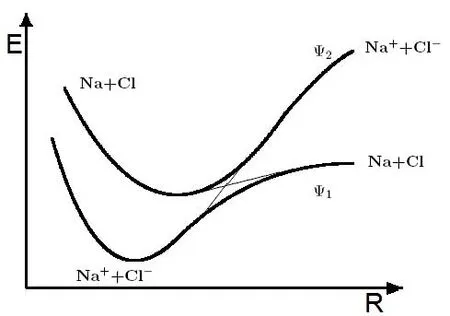
图1.NaCl分子的避免交叉势能面:粗线为绝热势能面;细线为交叉的非绝热态。图片来自[33]
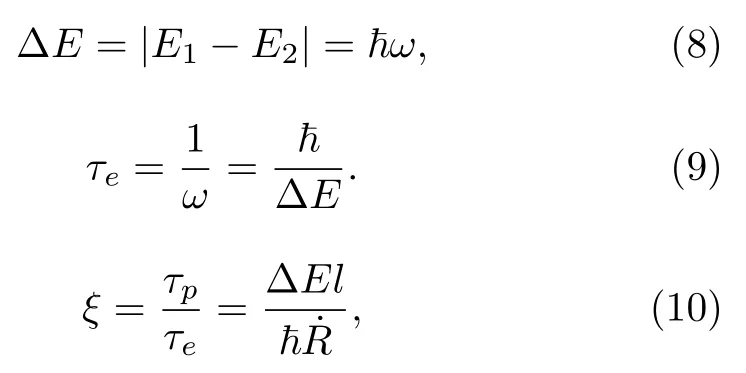
τp与τe之比ξ被称为梅西参数(Massey parameter)[24,35,36]。当时,也就是当能隙∆E较大并且原子核速度较小时,非绝热效应可以忽略不计。在这种情况下,绝热近似有效,体系一直在某一纯绝热态k上演化,即|ak(t)|2=1,方程(6)一直为0,即k=0。总电子波函数方程(4)只包含了一项,因为体系只涉及一个电子态,即:
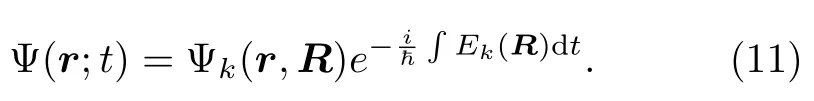
原子核的演化可以通过求解牛顿运动方程:
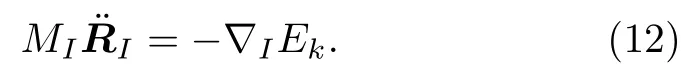
III.非绝热分子动力学
A.Ehrenfest方法
Ehrenfest方法又称为平均场轨迹方法 (meanfi eld trajectory method)、自洽经典路径法.selfconsistent classical-path method)、半经典含时自洽场方法 (semiclassical time-dependent self-consistent-field method)[37]。不同的命名意味着对细节的处理不一样,例如初始条件和边界条件的不同,但是运动方程的本质是一致的。
这里考虑的非绝热性也涉及到了绝热态分布几率|ak(t)|2,不同点在于|ak(t)|2不是一个常数,它会随着原子构型的变化而变化,即|ak(t)|21。显然,变化的|ak(t)|2会引起电子云发生形变,电子云的变化同时又会影响原子核的轨迹。尽管有些情况下电子非绝热性对原子核运动的影响微不足道(例如:高能的碰撞或者两个绝热态间能隙较大),但是在许多化学体系中,正确地处理电子-原子核之间的反馈是十分重要的[38,39]。最简单的方法是用能量期望值Eeff取代(13)式中的绝热势能面Ek,

因此,原子核就会在这样的有效平均势能面上演化,而有效势能面的获得又是通过绝热态以及它们的权重|ak|2决定,这就是Ehrenfest方法的本质。在绝热近似中使用Ehrenfest方法是一种本质的非绝热处理。但实际处理中,Ehrenfest方法一般按照绝热近似下获得的绝热波函数展开。因此,Ehrenfest方法有一定的局限性:不同态对应的经典关机区别不太大。在某些情况下,不同态对应的经典轨迹相差甚远,Ehrenfest方法的平均势能面会带来非物理的结果。
原子核所受力的大小由方程(14)的梯度决定,
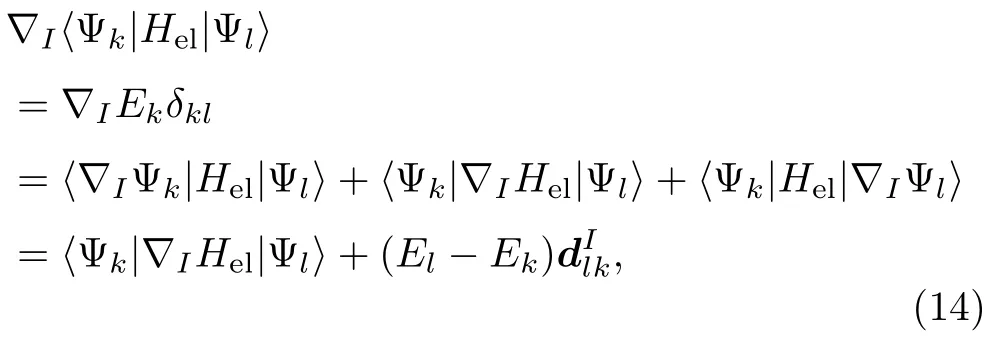

利用方程(5)和Hel的厄米性可知:
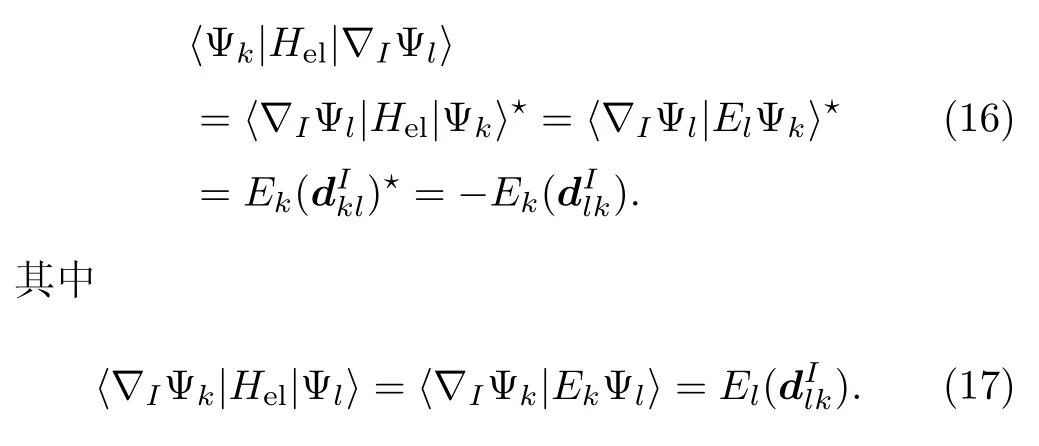
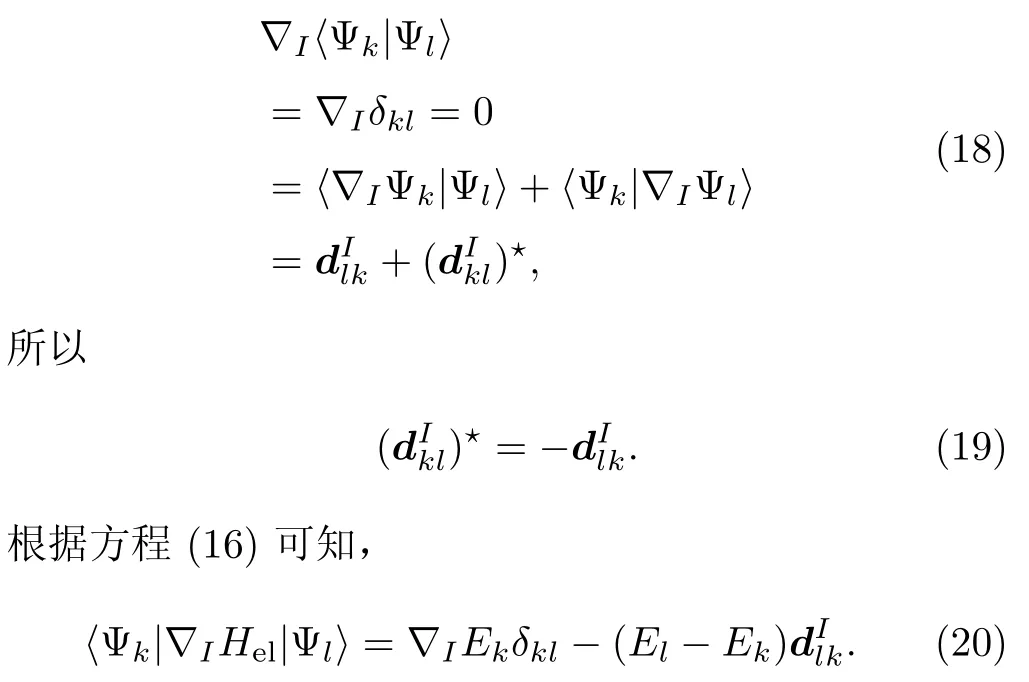
因此,方程(15)原子核受力的大小可以写成:
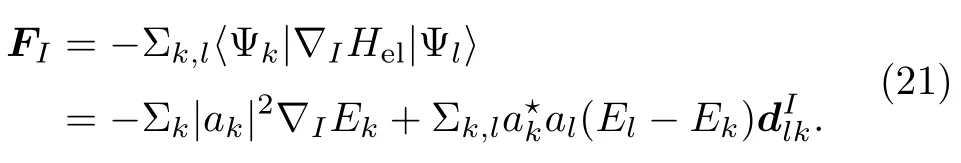
方程(23)说明了原子核受力来自两方面的贡献:第一项很容易理解是所有绝热态权重平均后的结果;第二项考虑到了绝热态占据数的非绝热变化。需要强调的是,原子核受力中的非绝热贡献的力的方向与非绝热耦合矢量一致。
原子核受力方程(15)的具体求导形式可以通过许多方法得出,例如:WKB近似[40]、程函方法(eikonal method)[41]、(半)经典含时自洽场方案[42~45]、密度矩阵法[46,47]以及经典极限的代数量子化[48]。以上不同的求导方法会导致Ehrenfest方案略微的不同。
Ehrenfest方法解决了许多化学问题包括金属面上的能量转移[49]、激发态寿命和有机分子的衰变特性[50]。但是由于它的平均场特性,该方法也存在许多严重的缺陷。初始时刻体系在一个纯的绝热态上演化,但是经过强非绝热耦合后,体系将会一直处于混合态。一般而言,即使是在位形空间 (configuration space)的渐进区域,波函数的纯绝热态的特性也难以恢复。当绝热态势能之间的差异显著时,平均势能的使用显然不能恰当地描述任何一个反应道,特别是研究一个占据数很小的反应道。通过Ehrenfest方法得到的只是一个偏离了“真实轨迹”的平均轨迹。
值得注意的是Ehrenfest方法不严格要求总波函数按照(绝热)基函数方程(4)展开,波包Ψ可以直接通过含时薛定谔方程(1)进行数值演化,但是为了使人们更加直观地去理解,有必要让波包Ψ在绝热态上进行投影(即根据绝热基函数展开)。对展开系数ak的研究是进一步优化非绝热方法的一个重要突破口,例如下面要介绍的面跳跃方法。
B.面跳跃方法
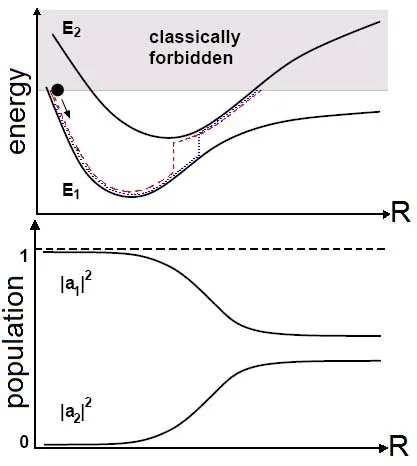
图2.上图:两个避免交叉的绝热势能面E1、E2,以及两种典型的面跳跃正反应轨迹(分别由虚线和点线表示)。非绝热跃迁几乎都发生在耦合区域内。下图:绝热态所对应的态占据数|a1|2、|a2|2。初始时刻体系处于Ψ1态并且动能为零,体系进入耦合区域后,Ψ2态的占据数增加。图片来自[51]
由上文的Ehrenfest方法可知,当体系通过并退出非绝热耦合区域进入渐进区域后,原子核的运动仍然受支配于所有绝热态的混合权平均,这是没有物理意义的。 因此希望用一个纯的绝热势能面描述渐进区域里体系的演化,这就是面跳跃方法(surface hopping approach)的基本理念。它不同于只计算一个“最佳”(态平均)轨迹的Ehrenfest方法,面跳跃方法涉及了一组轨迹(即大量的路径)。任何时刻体系总是在某一个纯绝热态上演化,但是具体在哪一个绝热态 Ψk上取决于每个绝热态的占据数|ak|2。因此改变绝热态占据数|ak|2的大小可以导致不同绝热势能面之间的非绝热跃迁,见图2。在绝热态k上演化的轨迹的平均个数永远等于绝热态k的占据数|ak|2。
值得注意的是,有三种方式可以实现面跳跃:1、基于 WKB波函数关联的半经典方法[52~63]。2、随机实现一个确定的多态微分方程(例如,量子-经典刘维尔方程(Liouville equation)[64~66])。3、准经典模型如众所周知的Tully等人提出的面跳跃法[67~81]。本文讨论的面跳跃法是最后一种Tully型面跳跃法,因为它已经成为处理非绝热光动力学最流行的方法之一。
在面跳跃方法的发展史中,Tully和Preston[25]两人最初的设想是绝热态之间的跃迁只能发生在某一特定的位置区域,而且这个位置区域在模拟计算之前就设定了。Tully[26]后来将非绝热跃迁发生的位置推广到整个区域,也就是非绝热跃迁可以发生于位形空间的任意一点。同时,Tully也提出了最少面间跳跃法(fewest switches algorithm)[26]。其基本思想是:在保证整体平均态占据数正确的前提下,每个轨迹只发生最少次数的面间跳跃。之后,最少面间跳跃法得到了广泛的应用,因为频繁的面间跳跃导致的结果就是雷同于Ehrenfest方法:所有绝热态的权平均。
因为面跳跃过程涉及了电子与原子核自由度的耦合,它首先要解决的问题是:如何建立良好的面间跳跃的标准和如何调整动量。现在我们来推导最少面间跳跃的标准。设体系总的轨迹个数为N,在t时刻时有Nk个轨迹处于Ψk态,
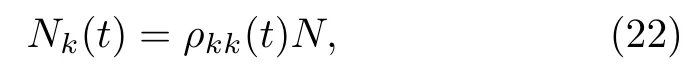
这里我们引入密度矩阵ρkl,
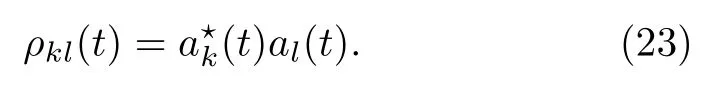
δt时间之后,即态上的轨迹数变化为:

在时间间隔[t,t+δt]内,一个轨迹从Ψk态跃迁到其它任意一个态上的几率Pk(t,δt)为:
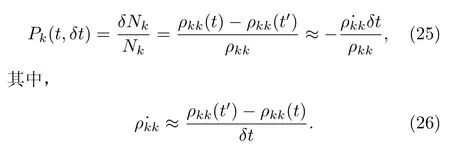

将方程(6)代入上式可得:

再将上式代入方程(27)中可得几率Pk的表达式:

轨迹从Ψk态跃迁到其它态上的几率Pk同时也等于轨迹从Ψk态跃迁到某一特定Ψl态的几率Pkl之和,即

由方程(31)和(32)可知:

轨迹从Ψk态跃迁到Ψm态成立的条件是:

ζ(0≤ζ≤1)是均匀随机数,并且是轨迹从Ψk态跃迁到前m个态的跃迁几率之和:

为了保证面跳跃之后体系总的能量守恒,应重新调整原子核的速度。通常的做法是只改变非绝热耦合矢量dkm(R)方向上的速度[26,60],见方程 (17),其本质与 Pechukas力相关[82]。在 Ehrenfest方法中,非绝热耦合对力的贡献也与非绝热耦合矢量dkm(R)的方向一致,见方程(23),这定性地说明了调整原子核速度的做法是可行性的。当然,这样做会导致原子核速度的不连续性,这正是面跳跃法的一个缺陷。在大多数情况下,非绝热面间跳跃只发生在两个绝热态间能量间隔很小的位置,所以对原子核速度做的调整也是相当小的。不过,面跳跃方法的一个严重的缺陷是无法处理所谓的经典禁戒跃迁 (classically forbidden transitions),即动能太小不足以补偿势能面之间的差值。Tully针对这种情况最初提出的建议是不执行面间的跳跃并且保持原子核速度不变,此方法被证实比之后的一些办法更准确[60,67,77]。图3阐明了经典禁戒跃迁是如何导致占据数|ak|2与k态上演化的轨迹数的实际百分比之间存在差异的。
同Ehrenfest平均场方法一样,面跳跃法也不能满足微观可逆性。这意味着这些过渡态理论在处理小概率事件时是不严谨的[83],因为在过渡态状态下半数的轨迹都必须及时地被统一为正向或逆向反应。
不同于 Ehrenfest方法的是,面跳跃方法依赖于表示形式 (representation-dependent),这一点已被证实[39],本文选取的绝热表示形式最适于模拟面间跳跃。
值得注意的是对方程 (6)沿整个轨迹积分发现面跳跃法存在很大程度的电子相干性。一方面,这使得该方法能够重现量子干涉效应[26]例如斯特科贝尔克振动(Stuckelberg oscillation)[24];另一方面,因为用经典力学来描述原子核的运动,电子自由度退相也相对过慢,这是 Ehrenfest方法和面跳跃方法共同的缺点。许多合并了退相干的半经典方法被提出[58,68,84~88]。在一定程度上,递推被自然地抑制了,但是增加了自由度的数量(例如在在凝聚态里)。即使是用气态小分子也可以证明这一观点[89~91]。

图3.上图:两个避免交叉的绝热势能面E1、E2,以及两种典型的面跳跃正反应轨迹(分别由虚线和点线表示)。非绝热跃迁几乎都发生在耦合区域内。 点线上的叉表示在经典禁戒跃迁区域跃迁被阻止。下图:绝热态所对应的态占据数|a1|2、|a2|2。初始时刻体系处于Ψ2态并且动能为零,体系进入耦合区域后,Ψ1态的占据数增加。体系退出耦合区域后,Ψ1态的占据数又减小。对于构型R,当E2处于经典禁戒跃迁区域时,Ψk态上的轨迹个数与总轨迹数百分比N不等于|ak|2;这里N为零,N保持为常数。图片来自[77]
总之,面跳跃方法至少可以定性地研究复杂的光诱导电子激发及振动激发及振动弛豫动力学,而且得到的结果总体质量比Ehrenfest的结果好。
C.连续面交换方法
Hack等人[68,87,88]为非绝热碰撞提出了一个新的半经典方法,它将面跳跃法的优点(在渐进区域体系处于纯态)和Ehrenfest平均场方法的优点(连续的势能同时避免无效跃迁)结合在一起。该方法是一种变形的含时自洽场方法,被称为连续面交换方法(continuous surface switching:CSS)。在强耦合区域,它与Ehrenfest平均场方法十分相似:不依赖电子的表示形式且自洽;在渐进区域,轨迹在纯态上演化。此外,该方法不存在跳跃、不连续性、以及跳跃受阻(frustrated hop)等缺点,并且总的能量和角动量都自动守恒。但是连续面交换方法不能够克服面跳跃法和Ehrenfest法共有的毛病:不能精确描述深层量子(deep quantum)过程,可是与其它已有的混合量子-经典方法相比,它又更加有用。
连续面交换方法涉及了一个对称的K×K非绝热势能矩阵且矩阵元为Ukk′。对于两个分子碰撞的情况,Ukk′矩阵元里的k 6≠k′在渐进区域将会消失,导致了渐进区域里的绝热与非绝热表达形式一致。
体系采用Meyer和Miller[92]两人提出的经典模拟哈密顿量(classical analog Hamiltonian)HCA,也就是将局量子电子态和经典原子核路径这两个矛盾体通过这个虚构的哈密顿量改为一种等效的经典问题。
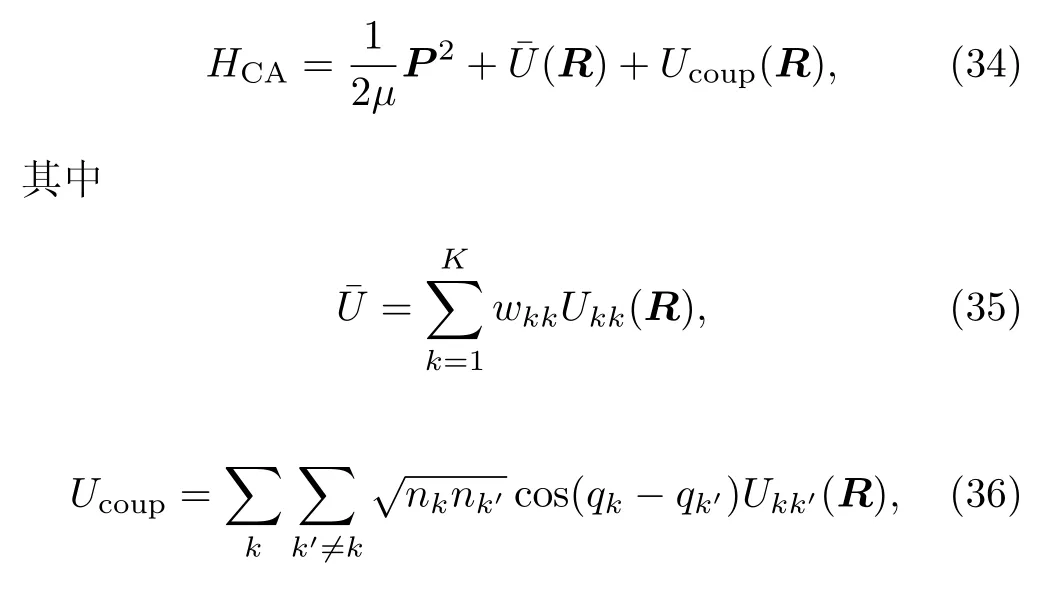
µ是约化质量,R和 P为原子核的坐标与动量,qk和nk分别为电子的广义坐标和广义动量,并且
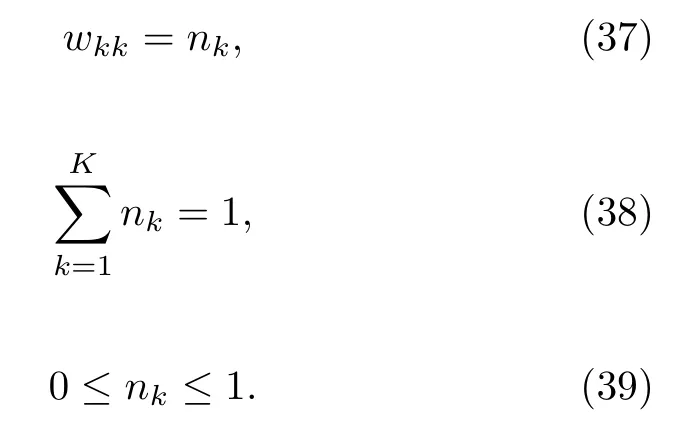
Ehrenfest方法的主要缺点是难以实施正确的边界条件;当Ukk′=0时,在权平均势能面上演化的体系也趋向于终止了在混合态的演化[92~96],因此会产生非物理的末态。至今没有一个完美无缺的方案可以解决这个边界条件问题。尽管继续研究边界问题是可行的,但很明显,任何一种涉及了双头边界条件 (double-ended boundary condition)的方法比只包括了量子化的初始条件的标准[97]准经典轨迹方法更加复杂。这里的连续面交换方法采用了其它的方法避免产生混合态的问题并且不牵扯双头边界值的问题。
该方法的精髓是用不同的方式来定义U。U函数为原子核运动的有效势能:当绝热势能面强耦合时,原子核坐标和动量的演化以Ehrenfest法的方式发生,当势能面不耦合时,体系又立刻在单一的势能面上演化。非对角元Ukk′自始至终保持了Ehrenfest的形式。电子的坐标qk只出现在Ucoup中。nk和qk的运动方程为正则方程:

方程(37)里的权重wkk被设为关于nk的函数,假设K=2,可得:
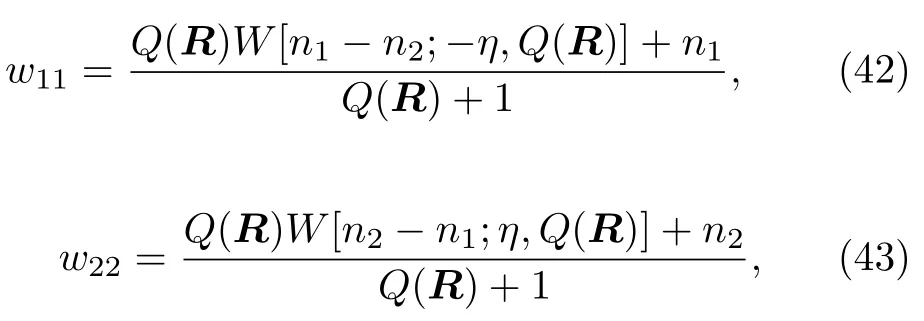
其中,η是参数(见下文)

方程 (46)中的W函数是海维赛德阶梯函数(Heaviside step function)Θ(x)的预设形式,
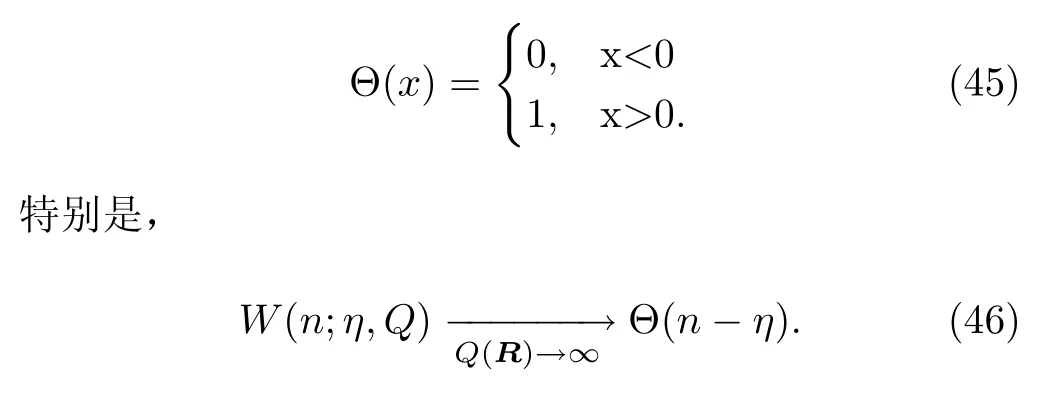
当Q(R)→∞:n1-n2+η>0时,w11→1,w22→0,体系在基态上演化;n2-n1-η>0时,w11→0,w22→1,体系在激发态上演化,这体现了面跳跃法在渐近区域的特点。 当Q(R)→0:w11→n1,w22→n2,呈现了Ehrenfest方法的特点。将方程(44)和(45)相加并代入方程(46)得:

尽管上式成立,也不能将方程(44)替换为w11=1-w22,因为n1和n2通常都是独立变量,但是当Q为零时必须w11→n1克服Ehrnfest方法的限制。
关于Q函数的选取,应满足:在强相互作用区域趋于零;而在非耦合区域趋于∞。

其中U0为参数。在强相互作用区域,∆Ukk′项的值很大,则Q值趋于0,对应了Ehrenfest演化;相反,在相互作用区域之外,∆Ukk′值趋于0,则Q值变得非常大,对应了面跳跃演化,体系依赖η值得选取,光滑且连续地从一个态切换为另一个态。(关于∆Ukk′,η和U0详见文献[68]。)
用MXH模型体系来评估这个CSS方法,M是金属原子,H是氢原子,X是非金属原子,M∗表示金属原子处于激发态。
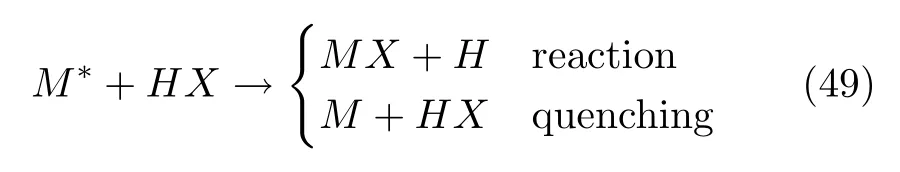
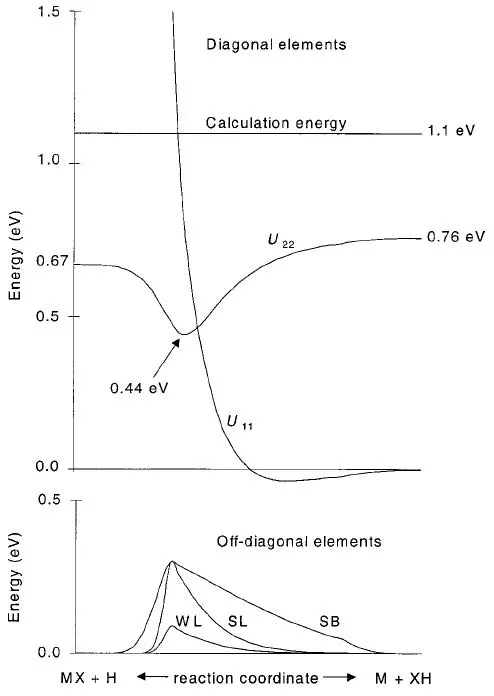
图4.非共线反应(∠≈120◦)中非绝热势能的示意图。反应物和淬火产物在右侧,反应后的产物在左侧。横线表示Etot=1.1 eV。图片来自[68]
图4给出了非绝热势能里U11、U22以及U12=U21的示意图。其中非对角元为:

r表示两个原子间的距离。A12决定了非绝热耦合的最大值,A12较大时,用“S”表示强耦合,反之,用“W”表示弱耦合。β12,2控制耦合宽度,用“B”表示耦合区域宽,“L”表示只有局部区域耦合。图4中下图分别给出了SB、SL和WL三种不同的势能面,具体的参数见表I。需要指出的是Truhlar方法参数化了处理过程,但各种参数视不同的情况而定。

表I.不同参数的取值[68]。
Hack等人在原始CSS方法的基础上进一步提高算法得到CSS2方法[87]。CSS2和CSS一样尽量将Ehrenfest法和面跳跃法的优点结合起来处理电子的非绝热过程[39,69],并提出连续面交换法需满足9个条件:

CSS方法只能满足前 5个条件,而 CSS2方法能满足所有条件,并且比 CSS和面跳跃法更加精确,此外,CSS2选取的函数更加简洁高效。在CSS2方法中,
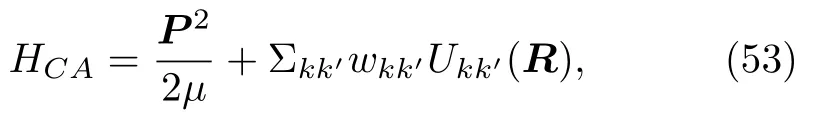
只是权重表达式变换为:
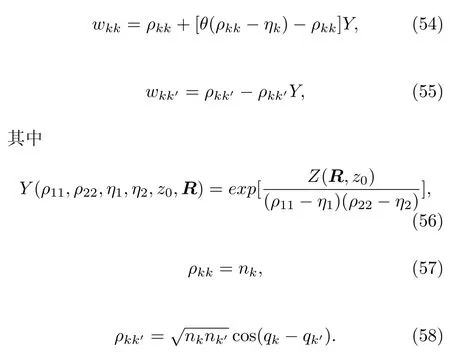
关于CSS2的细节见[87]。此外,还有许多关于将面跳跃方法和Ehrenfest平均场方法优点结合的方法[98,99]。
D.混合量子-经典刘维尔分子动力学
无论是经典体系还是量子体系均可用刘维尔方程 (Liouville equation)描述。在量子力学中,刘维尔方程为[100~102]:


N为体系的自由度个数。体系中任意一个变量也可定义为:

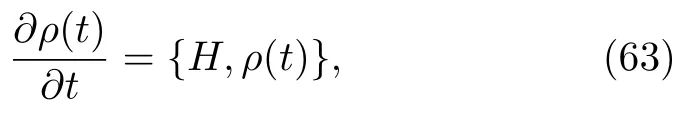
其中,ρ(t)为 Wigner密度函数,H为经典哈密顿量,同时,ρ和H为经典相空间变量 (q,p)的函数。是经典泊松括号 (classical Poisson bracket)。
量子刘维尔方程(62)和经典刘维尔方程(65)相似的表达形式促使许多理论研究者去构造一种混合的量子-经典刘维尔方法(mixed quantum-classical Liouville method:QCL)[48,64~66,112~125]。因此该方法对重粒子采取了部分经典极限,却保留了轻粒子的量子力学特性。算符ρ(t)和H涉及到电子自由度的部分通过基态|Ψni来描述,而涉及了原子核的自由度的部分用坐标q={qj}和动量P={pj}来表示,以ρ(t)为例:
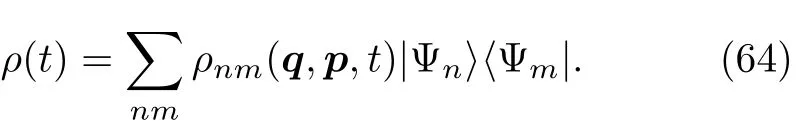
对应上述量子-经典密度算符的标准刘维尔方程为:
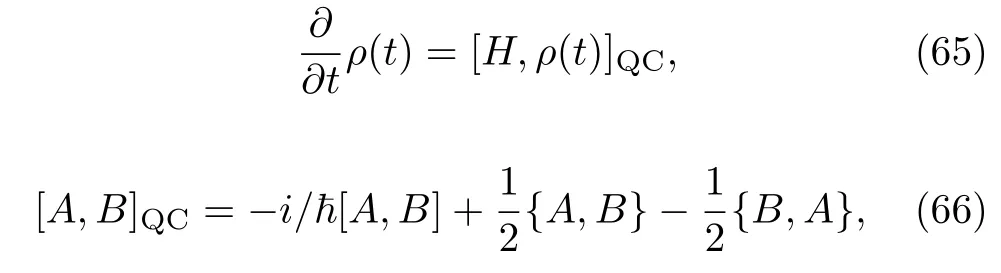
[·,·]QC记为量子-经典括号,简称 QC括号,它包括了方程 (62)中的括号 [·,·]和方程 (65)中的经典泊松括号{·,·}。 许多不同的方法都可以推导出方程 (67)和 (68),例如:Martens等人利用泊松括号和原子逆转换括号代替原子核转换括号[115]; 或者对 Wigner相空间表示实施部分经典极限[114,117,118];又或者对路径积分的影响函数(influence function)进行向前向后的线性化[125]。
尽管不同方法中的QC括号是不同的,但是整体上仍存在一致性[48,114,117]。QCL描述似乎是一种很有前途的方法,它自动准确地解决了电子的相干性和与电子跃迁导致的动量改变问题,并且消除了混合量子-经典方法中内部矛盾(即量子描述与经典描述的对立性)。此外,对于二次哈密顿量,经典Wigner近似是精确的,因此,对于线性振动耦合哈密顿量和大批涉及了锥形交叉以及旋转玻色子的模型,QCL方程也是精确的[37]。
后来的实际运用显示QCL方程的一般解法需要耗费与精确求解量子力学相等的工作量。只有相空间密度用网格形式表示时,才简化了偏微分方程(67)的数值实现。网格在相空间中可以是固定的[115],也可以是移动的,例如通过高斯波包[65,116,121]。同全量子计算一样,QCL方法的计算量随量子的和经典的自由度数量增加呈指数增长。为了得到能直接适用于真实多维体系的数值方法,需要采用包含了局域经典轨迹蒙特卡洛采样的随机方案[64,66,122~124]。但这又会面临两个难题:非局域相空间算符的表示和采样程序中的收敛问题,而复数的轨迹和快速振荡相使问题更难处理。其中采样收敛与所谓的动态信号问题有关,例如众所周知的实时路径积分方法[126]和含时半经典方法[42]。此外还有ANDO K等人也提出了一些不同的形式。
前面介绍了混合量子-经典刘维尔方法的基本思想,这里着重讲解它的理论和数值实现。对经典的原子核变量实施部分Wigner转换后,分子哈密顿量为:
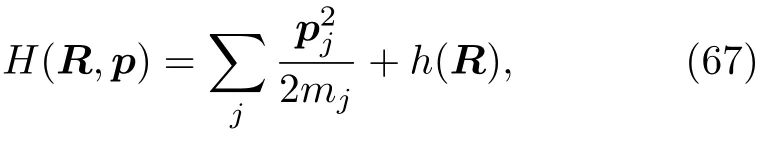
其中,原子核坐标R和动量p是经典的自由度,h(R)=Tq+V(q,R)是电子的哈密顿量,它涉及了电子的坐标q。如果选用绝热的电子表达:基态电子哈密顿量h(R)变为对角的形式:
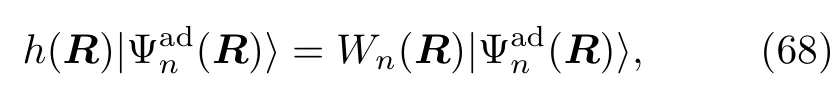
Wn(R)是第n个绝热态所对应的势能面。.分 Wigner变换的密度算符ρ(R,p,t)所对应的电子矩阵元为:
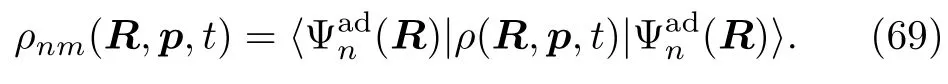
将方程(69~71)代入QCL方程(67)中,量子-经典密度矩阵的运动方程的绝热表示为[118]:
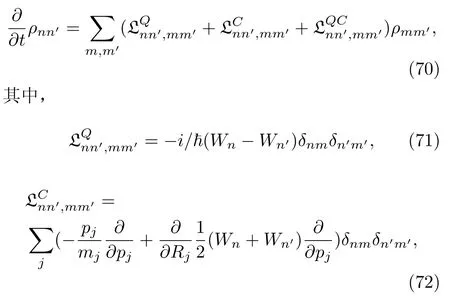
这两项分别体现了纯量子力学(Q)和纯经典力学(C)的密度矩阵随时间的演化。因此,量子部分 LQ为密度矩阵中的非对角元引入了相(Wn-Wn′)/,而经典部分LC针对绝热势能(Wn+Wn′)/2描述了标准的牛顿动力学。值得注意的是:运动方程(72)的演化需要经典轨迹的传播,这涉及了电子密度矩阵中的对角元和非对角元。这一点与传统基于波函数的面跳跃法相反。面跳跃方法不精确计算电子态间的相干性ρn6=m(t),它所需的经典轨迹的传播只涉及了密度矩阵的对角元ρnn。
刘维尔算符中的量子-经典部分为:
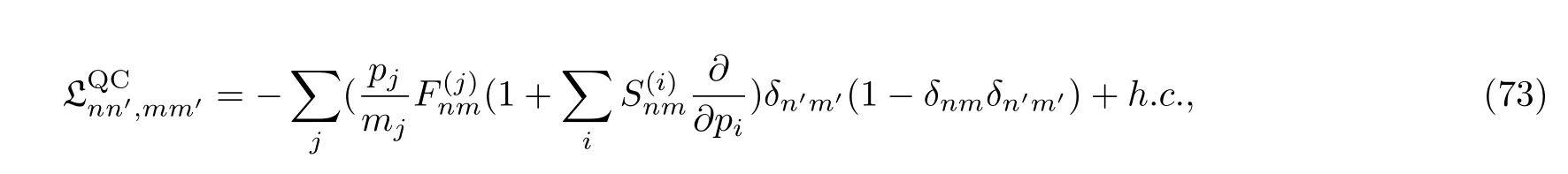
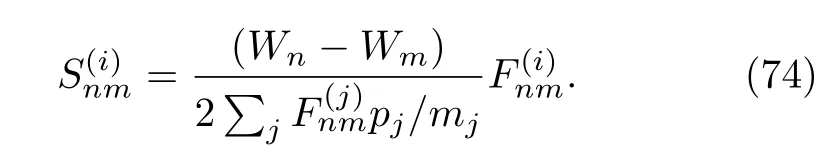
需要注意的是,在基于轨迹演化的QCL方程中,耦合项(75)对动量求数是困难的,因为该耦合项不仅需要相空间中某一特定点上的信息,还需要这个点周围区域的信息。一个可行的解决方法是将经典轨迹转演化变为高斯波包在网格中的传播[65,121]。Wan和Schofield两人[66]提出了一种替代的方法:同时对受力矩阵进行对角化消除矩阵中的非对角元,避开了前面提到的问题。但是这种替代的方法只适用于力矢量只有单一成分的一维体系。具体的混合量子-经典刘维尔方法参看文献[37]。
E.从头计算多次产生法
从头计算分子动力学 (ab initio molecular dynamics:AIMD)的目的是消除引言里提到的电子结构与分子动力学之间的界限。由于 AIMD方法和计算上的优势,它已逐渐成为化学物理中的主流方法。它需要同时处理电子薛定谔方程和牛顿方程,这一点再次强调了电子与原子核之间的紧密联系:电子支配着势能面的形式,而原子核又在这样的势能面上进行运动。1978年,Leforestier首次提出了AIMD的思想[127],但是量化计算部分特别繁琐复杂,这阻碍了AIMD的快速发展。幸运的是在过去的 25年里,Car-Parrinello (CP)方法[128]的引入大力推动了 AIMD的发展与普及[129~136。 CP方法的特点是运用扩展的拉格朗日量[137~142],但是并非所有的AIMD都采用扩展的拉格朗日量。有趣的是,CP方法的核心是电子结构与分子动力学之间的界线是模糊的。
大多数的AIMD将原子核看做经典粒子,因为多数情况下,牛顿动力学足以应付原子核运动。但是在定性研究中,原子核的量子力学性质都至关紧要时,例如:化学过程中的质子转移必须考虑到量子力学里的遂穿效应;此外,当绝热近似不再适用时,如:多重耦合电子态参与的化学反应,特别是有关锥形交叉的光化学反应,都必须考虑原子核的量子效应。本小结介绍的方法就是从头计算多次产生法 (ab initio multiple spawning:AIMS),它将同时求解电子和原子核的薛定谔方程。Multi spawing本身是一种波包的演化方法,可以在绝热或者绝热近似中表述。但是如果结合multi spawing和从头计算,很多时候是在绝热近似中才能工作,因为从量化中获得绝热态本身是一个很困难的问题[143~145]。AIMS将AIMD推广到可以同时处理电子和原子核量子效应的问题。AIMS用第一性原理分子动力学来描述光化学反应,包括了量子效应和非绝热效应,并且不引入经验参数。
体系的含时波函数由电子和原子核的波函数组成:
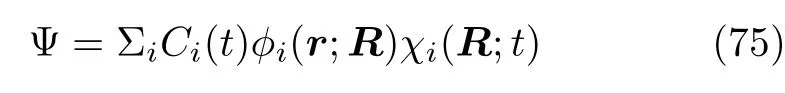
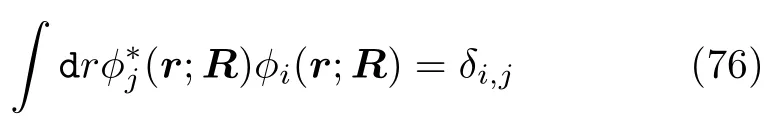
而含时原子核波函数χi(R;t)无需严格正交。 参数Ci为电子波函数振幅,它决定了电子态占据数|Ci|2和不同电子态间的相干程度,并可将它视为(约化)密度矩阵元:
两种电子基函数供电子波函数选择:一种是绝热基 (adiabatic basis),电子波函数的选取也是传统意义上的对角化哈密顿量寻找本征值,它非常依赖原子核的坐标R,并且处理非绝热的重任留给了算符;另一种是非绝热基(diabatic basis),它对原子核R的依赖性减弱。AIMS选用的是非绝热基,处理体系的非绝热效应,同时也简化了后面涉及到的算符。
AIMS通常使用薛定谔绘景(算符是常数,而量子态则随着时间演化),让时间效应体现在量子态上。再加上电子的波函数为非绝热基。因此,方程 (61)中的Ci(t)和χi(R;t)体现了体系对时间的依赖性,用以处理时间的演化;φi(r;R)承担了非绝热效应,同时简化了算符。
因为电子的波函数的形式与时间无关,因此体系的含时薛定谔方程可以改写为:
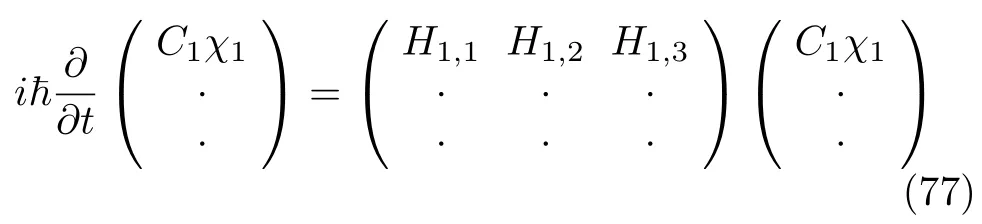
电子态的个数决定了上式矩阵的维度,哈密顿量包括了原子核的动能 ˆT(算符)和势能:H=T+V。以一维运动为例,原子核质量为M,则动能算符为:
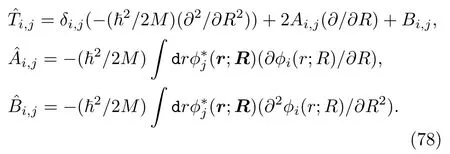
对应某一个(ith)电子态:用一系列线性组合的移动高斯(travelling Gaussian)函数来表示原子核的波函数:

其中含时高斯基态根据ith电子态的势能演化。 电子的势能是否绝热取决于电子基组的选择。 通常,不考虑不同态上电子间的耦合时,原子核波函数的每一个部分独立地描述某一态上的运动,即是ith电子态的含时薛定谔方程(哈密顿量为Hi,i)的解。与不同电子态上电子间的耦合有关(见下文)。这里对移动高斯采取Heller的“冻结高斯近似(frozen Gaussian approximation)[147]”的形式。波函数的演化通常是由初始的波函数通过传播子进行波函数的演化;而冻结高斯近似是将传播子与高斯波函数结合来描述波函数随时间的演化,即

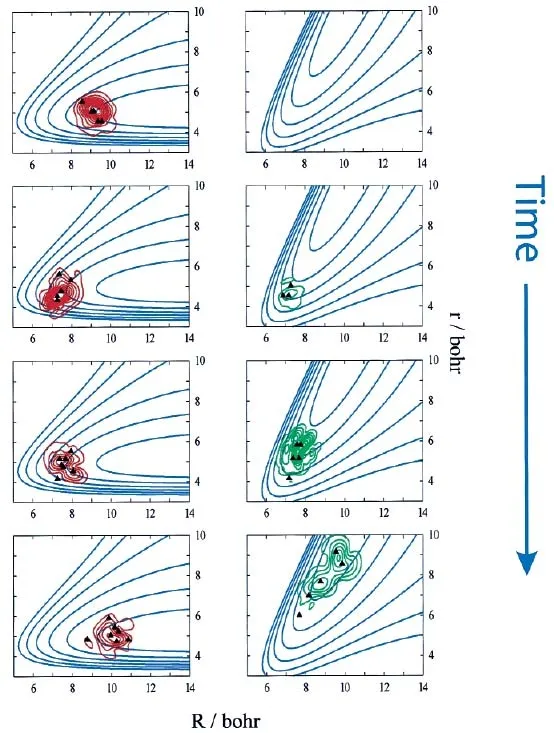
图5.共线碰撞A+BC→AB+C的AIMS原理图。左侧图和右侧图中的等高线(蓝线)分别为A+BC和AB+C所对应的非绝热势能面,采用雅克比坐标 (Jacobi coordinates):R为 A到 BC质心的距离,r为 B与 C之间的距离。重叠在等高线上的红线和绿线为原子核波函数的非绝热表达。计算始于单一的非绝热势能面上(左上图),当基函数穿过非绝热区域(原子靠近后返回)新的基函数会在另一个非绝热势能面上产生(spawning)(右中下图)。黑色三角表示单个高斯基函数的位置。图片来自[144]

冻结高斯近似的本质是:不同原子核基态之间的耦合包含了不同电子态上电子间的耦合 (interstate coupling)和相同电子态上电子内的耦合 (intrastate coupling)。

不同电子态上电子间的耦合包括了两项:
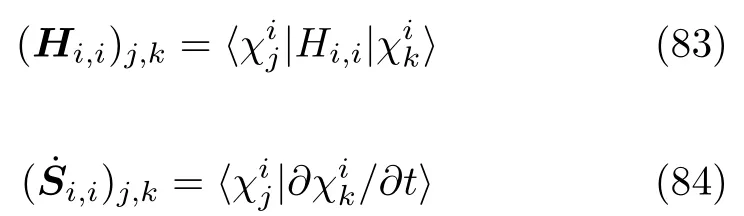
相同电子态上电子内的耦合至涉及了一项:

图5给出了A+BC→AB+C共线碰撞反应的原理图。图中的等高线为非绝热势能面由电子波函数决定,并且不依赖于时间,很明显与绝热势能面完全不同(见图6)。在模拟计算之前分别计算了不同势能面所能产生的轨迹,轨迹将牵引高斯基组中心的移动。体系随时间演化就体现于原子核的高斯函数在势能面上的移动。初始时刻,高斯基组只在对应的一个非绝热势能面上由相应的轨迹牵引演化,此刻只有相同电子态上电子内的耦合,没有不同电子态上电子间的耦合。当高斯基函数进入非绝热区域,不同电子态上电子间的耦合将增大,当它超过设定的阈值时,在另一个非绝热势能面上将产生新的高斯基组。这样就实现了从第一性原理出发描述一个非绝热原子核效应明显的化学反应过程。
总之,本节介绍了三类量子-经典混合的非绝热方法。第一类是首先得到一个量子力学精确的表达式(对整个体系的),之后对重粒子的自由度实现部分的经典极限。这个过程不是唯一的,它取决于具体的量子表达形式以及具体的经典极限方法。它主要包括了两种方法:Ehrenfest方法和混合量子-经典刘维尔方法。
对于Ehrenfest方法,由于平均场特性固有的矛盾,体系经过强耦合区域后不处于任何一个纯态上,因此不能准确地描述长时驰豫过程和多电子态过程。但是实际计算结果表明,最简单的平均场方法(MFT)比其它类似的方法能更好地描述复杂的非绝热动力学过程。例如含时自洽场方法(TDSCF)完全不能处理电子的长时间动力学行为,因为它忽略了自由度之间的所有关联[37]。
对于量子-经典刘维尔方法,计算结果表明它将有希望解决多维曲线交叉问题。密度矩阵的构造提供了一种自洽处理电子密度、电子关联和动量随电子跃迁变化的方式。尽管对局域经典轨迹采用了蒙特卡洛抽样,但是它仍然面临两大难题:非局域相空间算符的表示和相快速变化引起的抽样问题。虽然与其它的方法相比,量子-经典刘维尔方法在描述锥形交叉问题上精度和效率略差,但是它仍被广泛应用[64,65,116,121],说明该方法仍然具有开发潜力。
第二类是直接“结合方法(connection approach)”,该思想分别由Landau、Zener、Stckelberg提出。其中最流行的方法是由Tully等人创造的面跳跃方法。该方法能定性地研究复杂的光诱导电子激发、振动激发和振动驰豫等现象,并且计算结果优于Ehrenfest方法。但是它存在连贯性的问题,因此人们提出了各种各样关于跳跃算法的优化方法[68,71~73],不同的优化方法适合于不同的研究问题,因此不能简单地评价孰优孰劣。
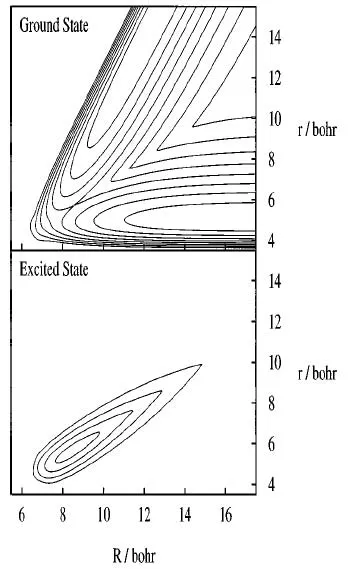
图6.等高线为与图9中对应的两个绝热势能面:基态和激发态。图片来自(145)
第三类是结合了Ehrenfest方法和面跳跃方法各自优点的杂化方法,这里只介绍了Hack等人构造的连续面交换法。该方法在大多数模拟计算中,能给出精度与全量子计算相媲美的结果。尽管半经典方法不能准确描述任意的一个反应道,并且由于量子结果的振动特性,半经典结果很难与它进行直接对比,但是CSS方法无疑是一缕曙光,它的结果有时更胜于全量子计算的结果。虽然CSS方法不是当前的主流选择,但是它的表现似乎在各方面都优于Ehrenfest方法,特别是在电子态间距离较大的体系。
本节最后介绍了能够描述原子核量子效应的从头计算多次产生法,该方法更加严格,所以计算量也相对较大。但是对于飞绝热矢量的处理是很困难的,有的用从头计算可以得出解析解,如CASSCF,有的则无法给出,这种情况就需要进行数值估算。还有在一些特定的情况下可以给出解析解,例如TDDFT。但是该方法仍有几个方面值得优化。首先,在电子结构处理上,应该逐步将分子的里德伯态考虑进来。其次,有待提高原子核动力学的精度。最后,需要注意其它的量子力学效应,尤其是零点能和遂穿效应等。
IV.EHRENFEST电子-离子关联动力学的应用
在20世纪末,TDDFT的发展与应用进入了黄金时期,例如用于研究分子、团簇149~156]、LiF和SiO2[157]、SiC[158,159]、石墨烯[160,161]、纳米管[162~164]在外场中的响应。
本节利用含时密度泛函理论 (time-dependent density functional theory:TDDFT)结合分子动力学 (molecular dynamics:MD)的方法,在局域密度近似 (LDA)下,研究了单一质子与单一甲醛分子非绝热碰撞过程中的电子激发、分子碎裂等现象。 TDDFT与 MD相结合的方法首次由 Saalmann和 Schmidt提出[165,166]:用含时密度泛函理论处理电子波函数的演化,大大减少了求解薛定谔方程带来的巨大计算量;用经典分子动力学处理原子核或离子实的运动。但是他们电子波函数采用的是局域高斯波,因此不能完全描述体系中电子的激发。开源代码PWTELEMAN[167]中提供的TDLDA-MD模型正好能弥补这一缺陷。
在碰撞体系中,将所有的粒子分为两类:一类是离子实(原子核与内层电子),看作坐标为Ri,i=1-Nion的经典带电点粒子;另一类是剩余的价电子,用单粒子波函数Ψn,n=1-Nel描述。在密度泛函理论中,所有可观察量均由电子密度的函数ρ=Σn|Ψn|2表示。则体系总能量可以写为以下形式:
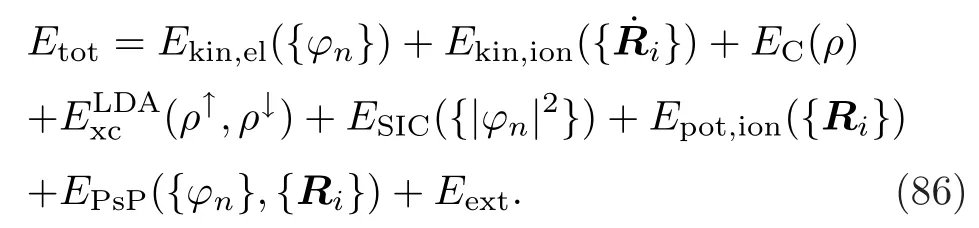
它包括了电子和离子的动能,电子之间的直接库仑能、交换关联能和自洽修正项,离子的势能,电子-离子间的赝势,以及外部作用于体系的库仑能。每一项都是关于ρ和R的函数。
含时Kohn-Sham(KS)方程主宰了电子结构的演化:
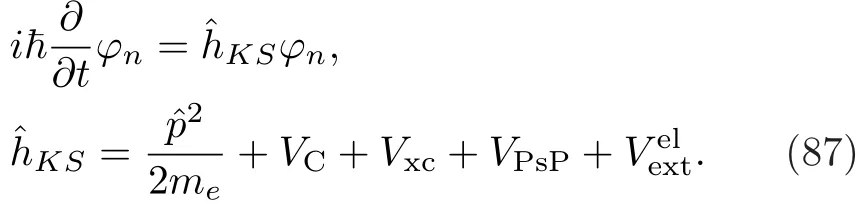
牛顿方程控制了离子实的运动:

TDLDA-MD方法同时处理了电子的演化和离子实的运动,提供了一种Ehrenfest型的非绝热耦合方法。当电子激发等现象可以忽略不计时,TDLDA-MD方法又会自动退化为绝热近似。采用的真实空间网格[168]有利于电子-离子间直接耦合。使用的吸收边界条件[169]有效地消除了体系中被激发的逃逸电子,体系中逃逸电子的个数为:其中是t时刻体系中的电子数,V为整个网格的体积。
TDLDA-MD模型已成功用于研究激光激发乙烯[170],质子与甲醛[171]、甲酸[172,173]、乙炔、乙烯[174,175]、乙烷[176]、甲烷[177]等小分子的碰撞。
A.乙烯在强激光场中的响应
给系统一个微小的激发后,系统的偶极矩做小幅震荡。对系统的偶极矩进行傅里叶变换后,可以得到乙烯分子的光学吸收谱,如图7所示。

图7.TDLDA-MD计算得到的乙烯分子光吸收谱。图片来自[170]
从图7可知,乙烯分子的光吸收谱中低频部分由独立峰组成,而高频部分则是连续平台。第一个谱带包含了在 7.74 eV处的最低峰和在 8.16 eV处的第二个孤立峰,这两峰均来自平行于 C=C双键的振动跃迁,它们与最高占据轨道(HOMO)π轨道-1b3u电子的激发相关联。第二个谱带对应 8.8~11 eV,它主要是由1b3g和1b3u两个轨道的电子在x、y、z方向的激发贡献的。
为了研究离子运动对乙烯分子在激光场中激发响应的影响,图8对比给出了电子-离子耦合关联动力学的计算结果与只考虑电子动力学而冻结离子自由度的计算结果。从图8(a)可以看出,在电子-离子耦合情况下,四个 H离子(实心方框表示)明显偏离平衡位置向远离C离子(空心方框表示)的方向运动。对于电子,从大约18 fs开始,电子开始逃逸,当激光场在100 fs关闭时,总共有2.8个电子被电离。从电子分布的均方根半径可以看出,从大约20 fs开始,rmsel逐渐增大,在经历几次振荡之后,当激光场达到最大幅度时,电子分布达到局域最大,之后尽管激光场逐渐减弱,但由于离子的运动及电离的共同影响,导致电子分布弥散更大,而且振荡更剧烈。
在图8(b)中,在离子冻结情况下,离子是固定不动的。对于电子,当激光场关闭时,总共只有2个电子被电离。比图8(a)中最后Nesc=2.8要小。这说明在考虑离子-电子关联动力学的情况下,分子的电离增强了。由于离子是固定的,在激光场达到最大幅度时,电子分布达到局域最大,之后,激光场逐渐减弱,rmsel开始作振荡。从大约70 fs开始,系统的电离趋向于饱和,电子密度开始趋向于新的稳定的分布。
以上两种情况下,分子在激光场作用下电离的不同及离子的运动说明了在研究分子在外场中的激发响应时,将电子、离子自由度耦合的重要性。有了这种耦合,我们能够探究不同参数的激光脉冲作用下的分子的激发响应。
图9给出了激光频率对乙烯分子激发的影响。这里主要考虑两种激光频率,一种为ωlaser=9.5 eV(图9(a)),它处于共振频率范围内,另一种为ωlaser=6.8 eV(图9(b)),它小于乙烯分子光吸收谱中最低峰对应的频率值(7.74 eV)。图9从上到下,分别对应乙烯分子键长、三个方向的离子均方根半径沿激光极化方向的偶极矩(Dx)及电离随时间的演化。
当激光频率处于共振频率范围内时,从图9(a)中可以看出,所有的 C-H 键 键长从 10 fs开始同步缓慢增加。当激光于 20 fs关闭后,.们继续同步增加。然而,从大约 25 fs开始,C-H键键长增加的同步性开始发生变化,具体表现为RC2-H3和RC2-H4比RC1-H1和RC1-H2增加的快。在 60 fs时,所有 C-H键键长已经大于 10 a.u.。这表明乙烯分子的四个 C-H键都已发生了断裂。对于C=C双键的键长(图中的实线),它开始增加的时刻要比C-H键键长增加的时刻要晚。整个100 fs过程中,它呈小幅振荡,这表明C-C键没有破裂。
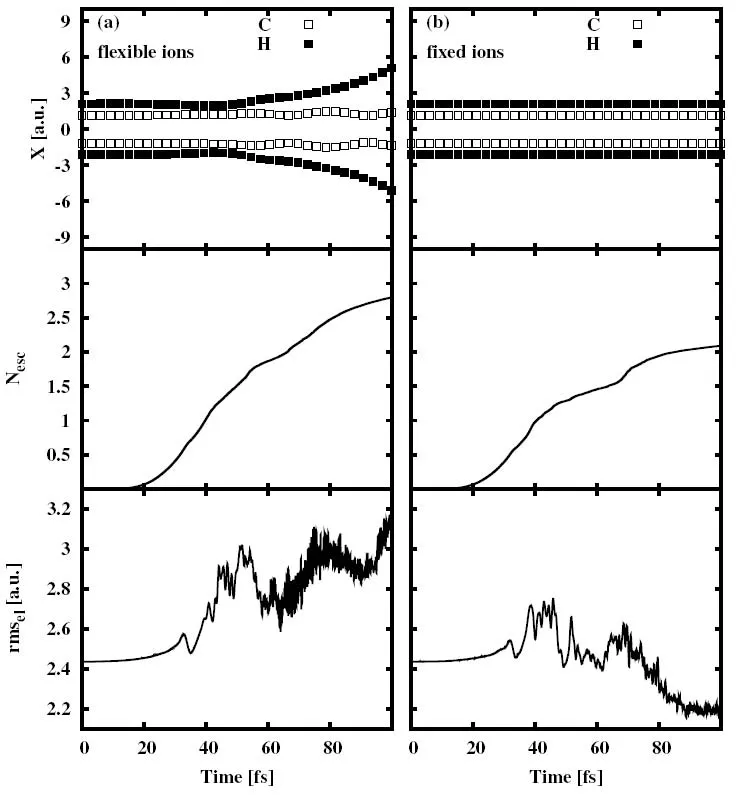
图8.离子运动对乙烯在激光场中激发响应的影响。激光脉冲波形采用cos2型,极化方向沿x方向,电场强度I=1013W/cm2,激光频率ωlaser=8.16 eV,脉冲长度Tpulse=100 fs。(a)和(b)分别对应电子-离子耦合动力学计算结果和冻结离子自由度的计算结果。两图中,从上而下分别是离子的x坐标、系统逃逸电子数、电子分布的均方根半径随时间演化图。图片来自[170]
对于离子的均方根半径,从图9(a)中第二个图可以看出,在x方向和y方向,从大约5 fs开始,离子的均方根半径表现出直接的增加,而在z方向,保持为零。这表明乙烯分子的运动发生在xy平面,而且体现出直接的扩张模式。而当激光频率ωlaser=6.8 eV时,即激光频率不在共振频率范围时,从图9(b)中的前两个图可以看出,乙烯分子的C-H键键长及C=C键键长作幅度很小的振动;在x方向和y方向上,离子的均方根半径随时间演化表现的是微弱的振动,这说明乙烯分子在xy平面内的运动是微弱的呼吸模式,而在z方向上,离子没有运动。
此外在激光强度、脉冲长度保持一定的情况下,当激光频率处于共振频率范围内时,分子的电离比激光频率小于共振频率时强,而且乙烯分子在其平面内发生了解离,所有的C-H键发生了断裂。
B.质子与甲醛分子碰撞
质子与甲醛碰撞的构型如图10所示:质子以 85eV的动能沿着平行于C=O双键的方向入射,且碰撞参数为b=0.3a0,即质子将穿越C-H键。

图9. 激光频率对乙烯分子激发的影响。(a)ωlaser=9.5 eV,(b)ωlaser=6.8 eV。激光的极化方向都沿x方向。两图中激光强度和脉冲长度都相同,I=1014W/cm2,Tpulse=20 fs。两图中从上到下,分别是乙烯分子键长、三个方向的离子均方根半径、沿激光极化方向 的偶极矩及电子逃逸数随时间的演化图。图片来自[170]
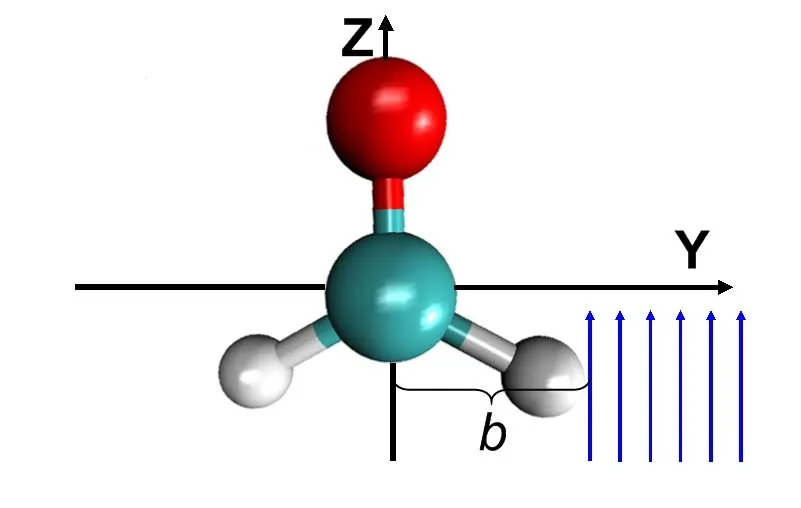
图10.质子与甲醛碰撞构型示意图,红球代表氧原子,绿球代表碳原子,白球代表氢原子,箭头方向为质子入射方向,b为碰撞参数:质子初始轨迹与甲醛质心之间的垂直距离。图片来自[171]
图111.出了该碰撞体系的演化过程中的几个重要时刻:t=8.47 fs时,质子离靶分子较远,因此对甲醛的影响也不大。t=12.1 fs时,质子完全进入甲醛分子中,并使它发生了轻微形变。t=13.31 fs时,靶分子中的OCH使质子反弹并与碰撞C-H键中的H原子碰撞,将部分的动能传递给了该H原子。于是质子有充足的反应时间去俘获电子,在t=14.52 fs时,质子与H原子各自带着少量电子欲离开体系。t=15.73 fs和t=16.94 fs充分展示了体系最后的C-H键断裂以及质子俘获电子离开的过程。
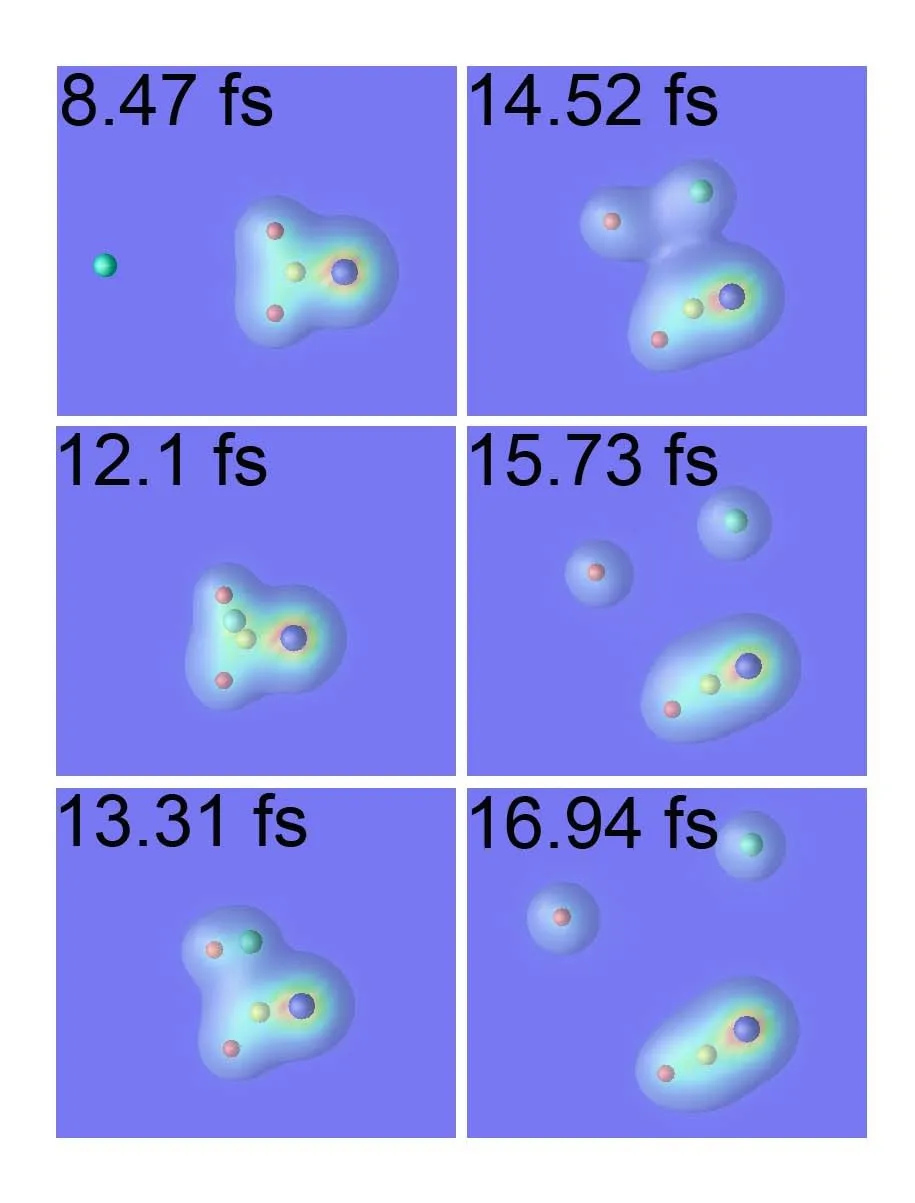
图11.利用TDLDA-MD模型计算碰撞参数为0.3a0时,85 eV的质子与甲醛碰撞反应的时间演化。
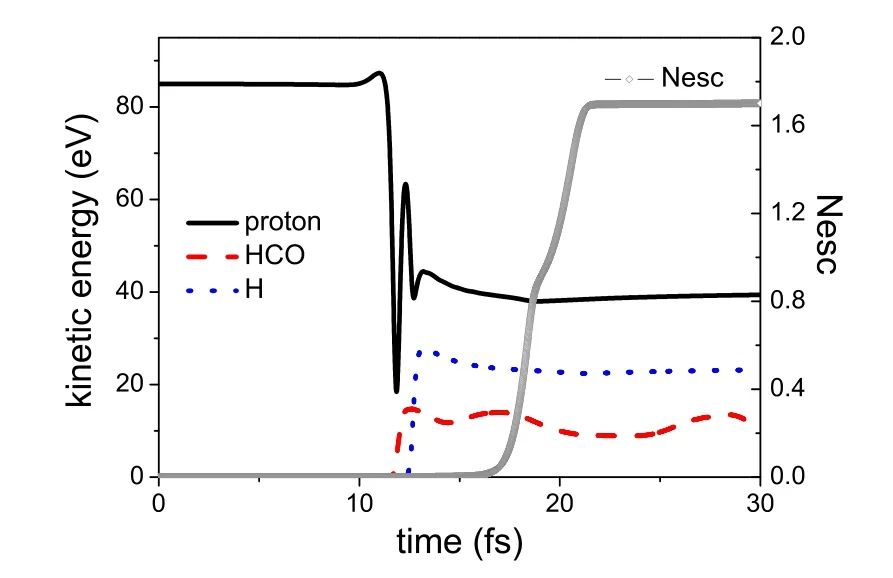
图12.85 eV质子与甲醛碰撞反应中前30 fs内各个碎片的动能变化以及体系总的电子逃逸数(Nesc)变化。左侧刻度对应动能的变化,右侧刻度对应Nesc的变化。
图12.出了反应中前30 fs内质子,保留的OCH以及逃离的H原子的动能变化,同时给出了体系的电子逃逸数的演化。初始时刻,只有质子运动且动能为85 eV。10 fs后,质子受到甲醛分子微弱吸引被加速,但是因为质子与靶分子的离子实发生了碰撞,排斥力立即取代了引力,质子动能急速下降,最后被反弹在排斥力的作用下加速。质子的远离又使得排斥力转换为吸引力而又做减速运动。在10 fs~13 fs之间,体系完成了绝大部分动能的转移。显而易见的是OCH碎片虽然最先获得动能,但是它只获得了大约15 eV,而H原子之后获得了约24 eV的动能。质子最后只剩下了约40 eV的动能,其余的6 eV能量全部转化为电子的动能或体系的内能,才导致电子的激发和逃逸。体系总的电子逃逸数为1.7。

表II.不同碰撞能量下的甲醛分解几率R(%)和电离截面σ(×10-16cm2)。σs:单电子电离截面;σd:双电子电离截面;σt:总电离截面。
当改变质子的入射能量,从表II可知,随着碰撞能量的升高,甲醛分子的分解几率明显下降,但是质子碰撞导致甲醛的各种电离截面均有明显的增加。
C.质子与甲酸分子碰撞
用动能为5 eV的质子与甲酸分子相互作用。由于甲酸分子的平面结构,故采用两大类碰撞方向(如图13所示):质子平行于分子平面内的入射,记为“A”,质子垂直于分子平面的入射记为“B”。A1为质子沿C=O双键方向入射,A2沿羟基O-H键方向入射,A3沿C-O单键方向入射,A4沿C-H键方向入射;B1为质子垂直于分子平面碰撞在O1原子周围,B2为质子垂直于分子平面碰撞在H2原子周围,B3是碰撞在O2原子周围,B4是碰撞在H1原子周围。
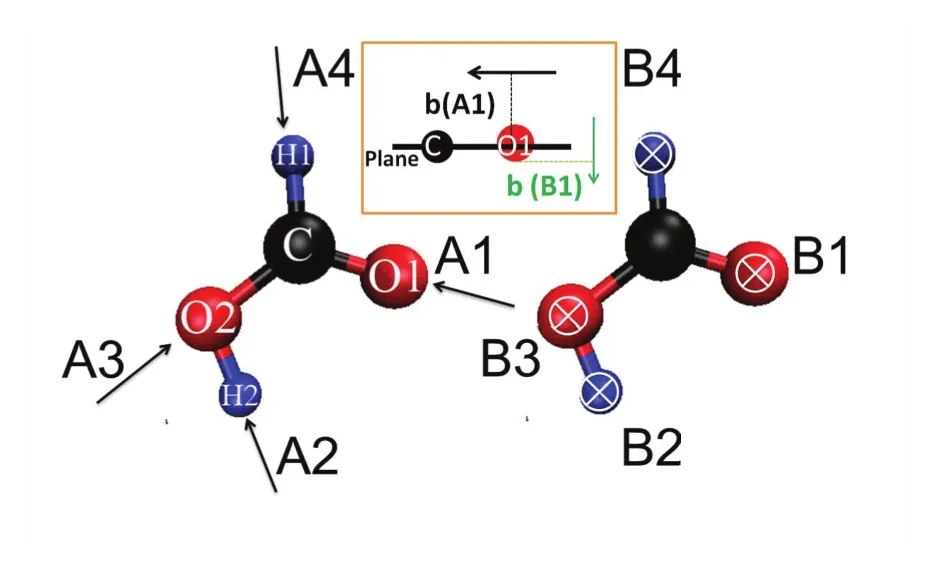
图13. eV质子与甲酸碰撞的 8种基本入射方向,插图以A1和B1为例给出了近碰撞的中碰撞参数b的示意图。图片来自(172)
E=5 eV时,当碰撞位点位于甲酸的氧原子上及其周围时(b≥0),如A1、A3、B1和B3方向,由于入射质子受到氧原子强烈的库仑排斥力而发生散射逃离靶分子体系。揭示了甲酸中氧原子具有屏蔽作用,能有效地保持了甲酸分子的稳定性。当碰撞位点在甲酸的氢原子及其周围时,如A2、A4、B2和B4方向,当碰撞参数较小时(0≤b≤0.6a0),质子被俘获与甲酸形成质子化甲酸分子(A2和A4)或者质子取代了被碰撞的氢原子(B2和B4)。这是因为质子与氢原子的质量相同,更容易导致动能从入射质子转移到被碰撞的氢原子上。而碰撞氢原子后是发生质子化还是取代的主要根源取决于相互作用时间和氢原子的运动方向。对于平面内碰撞的A2和A4,质子将能量传递给被碰撞的氢原子后停留,获得能量的氢原子开始运动但是其运动方向仍然是向着体系的其它原子,从而实现了分子内部的能量传递,并且总的动能很小,分配到每个原子上就更微弱了,因此体系就完成了质子化的过程。
E=2 eV时,甲酸质子化的发生几率将大幅增加,发现在A2、A3、A4、B2、B3和B4六种方向上均有甲酸质子化现象发生。说明降低碰撞能量能有效地提高甲酸分子质子化几率。

图14.E=5 eV时,A2方向上的正碰撞(b=0)下的电子离子随时间演化图,其中蓝色代表碳原子、黄色代表氧原子、红色代表氢原子、绿色代表入射的质子,电子密度越亮表示密度越大,且边缘轮廓的密度为ρ=0.01。图片来自[172]
同时发现了两种不同的甲酸质子化机制,以A2、A3和A4为代表的平面碰撞中,1,3-H迁移过程占决定性作用,如图14所示。从图中可知:当t=24.20 fs时,质子已经和甲酸分子很接近了,并且对甲酸的部分电子产生了微弱的吸引,使此刻的电子密度分布发生微弱形变。当t=30.25 fs时,入射的质子成功进入甲酸的羟基部分,导致电子密度发生巨大的变化,而且氢原子也发生了较小的位移。当t=36.30 fs时,质子的介入导致羟基中的H2原子被排挤到两个氧原子的中间区域,呈现了一个1,3-H迁移的现象。
1,3-H 迁移过程,对甲酸的质子化过程也意义重大。一方面甲酸的π-电离更加促进了 1,3-H迁移的发生,另一方面与打开 C=O键或 C-O键相比,入射的质子更容易打开 O-H键,因为甲酸和质子化甲酸中的各个键级不同(O-H键在 HCOOH中为 0.495,在 [HCOOH]H+中为 0.451;C=O双键在HCOOH中为1.344;C-O键在HCOOH中为0.726,在[HCOOH]H+中为0.937[180])。
当能量降低为 2 eV 时,在优势碰撞位点上 (A2和 A4),一次迅速的 1,3-H迁移即可形成质子化甲酸,在劣势位点上(A3),则需要两次1,3-H迁移,并且形成时间较长。
以B2、B3和B4为代表的垂直碰撞中,1,3-H迁移的作用被甲酸分子的出平面振荡削弱,两者共同决定了甲酸质子化的形成,如图15所示。因为甲酸分子是一个平面分子,它的出平面振动有助于打开原本稳定的C=O键,促成新羟基的直接形成。

图15.E=2 eV且b=0时,B2、B3和B4中整个质子化甲酸分子的各个原子的轨迹叠加图,其中黑色、红色、绿色、深蓝色和浅蓝色分别代表C、O、H+、H1和H2。图片来自[173]
在两种能量下,体系最终形成的都是稳定的羰基质子化甲酸,而不是羟基质子化甲酸,这与实验和其它理论的结论相符,因为甲酸分子中羰基氧的质子亲和力比羟基氧的质子亲和力大。 此外,在质子化甲酸中的 1,3-H迁移与分子出平面振动体现了氢的“floppy”无规律流动现象,该现象已经在其它质子化分子(和)中被广泛发现与研究。
D.质子与甲烷分子碰撞
碰撞能量为20~40 eV,选取了九个不同的碰撞方向,如图16所示。碰撞过程中,出现了多种反应通道:电荷转移,质子交换,碰撞解离,质子交换同时诱发解离,形成氢分子同时诱发解离等。甲烷碎裂的机制与方向有关,这是因为质子与甲烷的作用程度不同,能量的损失也不同。碰撞产物出现了与实验和END计算结果相一致的碎片类型,和分别占85.23%、13.5%和0.6%。这与实验测量的碎片比率不太符合,因为计算中认为每个碰撞方向是等概率的,这是一个非常粗糙的近似,为了符合实验的统计记过,可提高碰撞参数的分辨率或增加更多的方向。
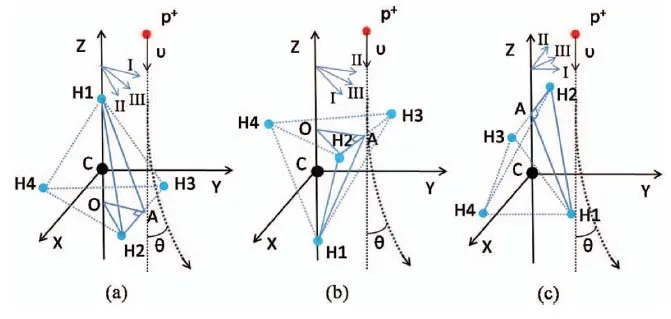
图16.质子与甲烷碰撞的9个不同方向。黑色、蓝色和红色分别代表碳、氢原子及质子。图片来自[178]
对于靶目标的离化目前有两种机制:电子激发和电子俘获。图17是质子进入 (14.52 fs)至离开(31.46 fs)计算区域的过程。第一次碰撞发生在16.94 fs,是由于电子受到了质子库仑力作用。当质子开始进入甲烷带电密度区域时(19.36 fs)碰撞出现,由于俘获电子数量远小于系统的总电子数,因此观测不是很明显。在20.57 fs和21.78 fs时注意到z=0处电子密度曲线变得尖锐,这可看作是质子携带俘获电子进入该区域的证据。质子继续沿着z方向运动,26.2 fs时密度峰又出现了,事实证明,质子碰撞过程中始终能俘获一些电子(29.04 fs)。当质子进入吸收边界时(31.46 fs),俘获的电子被吸收。对z方向积分可得,整个过程俘获的电子数是0.17。在图中总能在质子周围观察到一个小的密度峰,可以认为电子俘获是30 eV下甲烷离化的基本机制。
如图18所示,碰撞过程中,质子和靶分子间进行能量交换,这诱导了离子间的振动行为,在20.5 fs之前,所有的键长都处于平衡态,碳原子和质子的距离随着时间增加而单调减小。当质子距离靶目标最近时,C-H键中的振动被激发。由于H1靠近质子入射的路径,其振动是最剧烈的。由碰撞诱导的键长增大的现象叫“键稀释”效应。
散射角是碰撞参数b的函数,通过分析散射角来研究碰撞。图19为模拟碰撞和经典碰撞的散射角随碰撞参数b的图。可以看出在经典碰撞中,对心碰撞散射角接近180度,随着b增大散射角单调减小。而在实际的模拟中,在b增大到一定值时散射角出现一个极大值,即“rainbow”角,这是离子-分子散射的特征,是因为碰撞过程中散射角起源于库伦排斥力和吸引力的净作用效果。从图上看出计算得到的rainbow角为11.8度,实验值是10度,不完全符合的原因有两个,(1)方向效应。(2)理论描述。
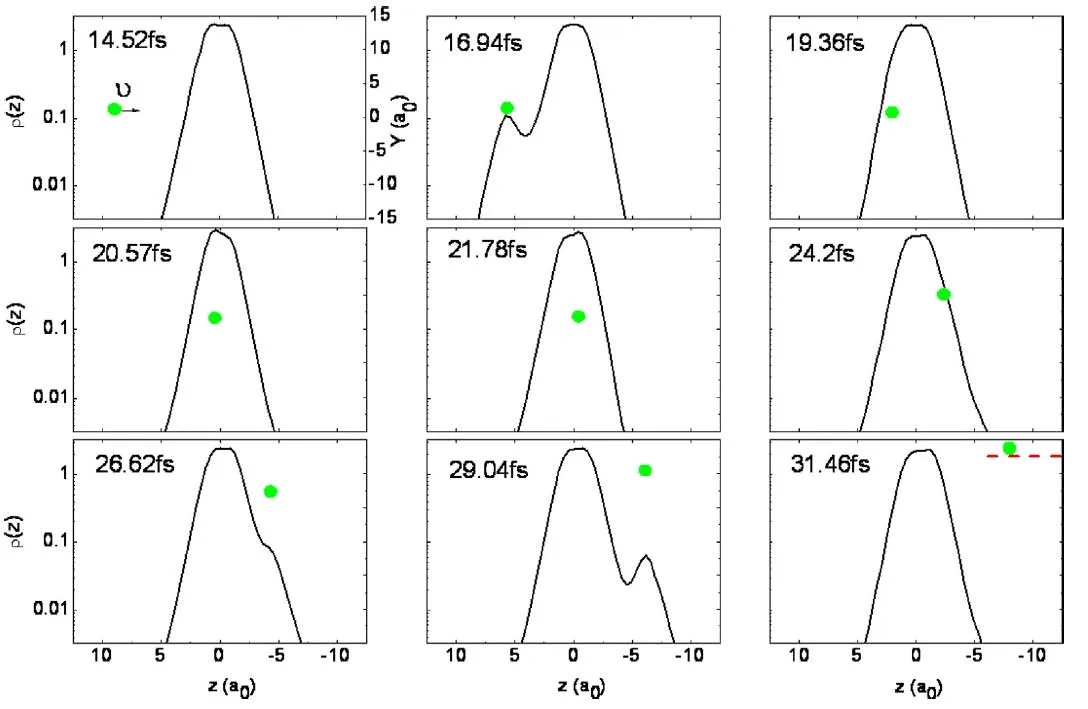
图17.30 eV质子与甲烷在方向edgeIII上的碰撞,b=0.9a0,电子密度在14.52 fs到31.46 fs的演化。绿色小球表示入射质子,坐标是双y坐标,左边采用对数坐标,表示电子密度,右边是线性坐标,表示质子在y方向上的位置。31.46 fs时,红色虚线表示边界。图片来自[177]
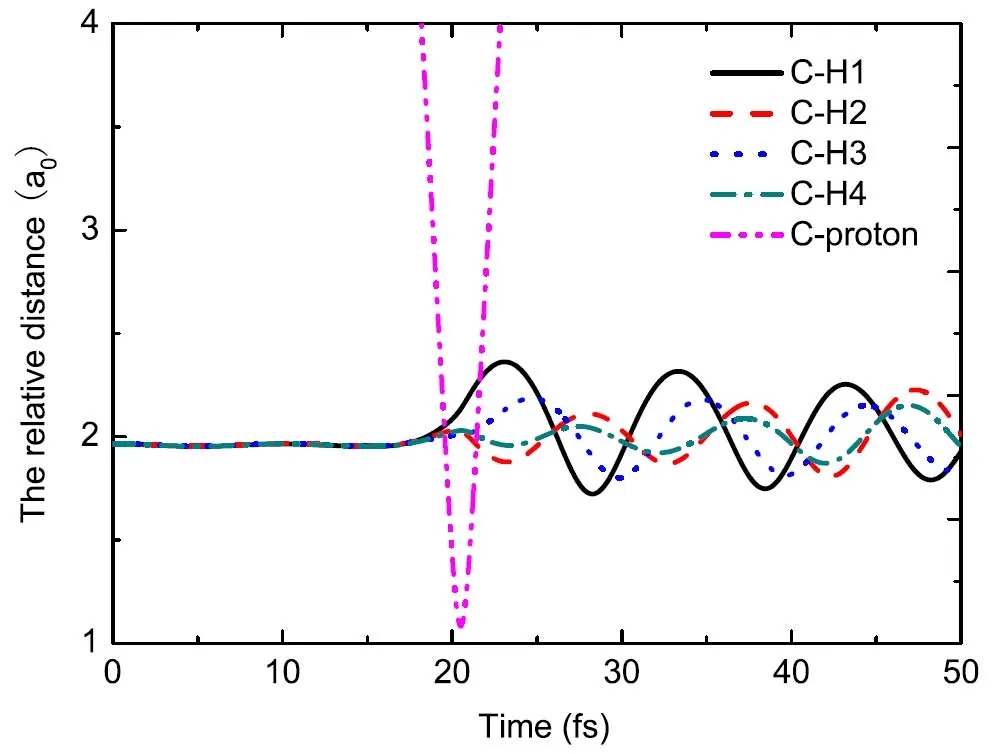
图18.30 eV质子与甲烷在方向edgeIII上的碰撞,b=0.9a0,原子间的相对距离。图片来自[177]
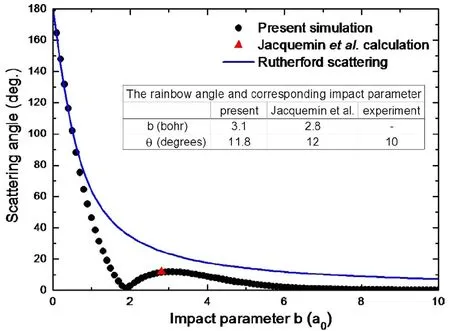
图19.散射角在30 eV能量下的分布。图片来自[177]
E.质子与乙炔、乙烯和乙烷分子碰撞
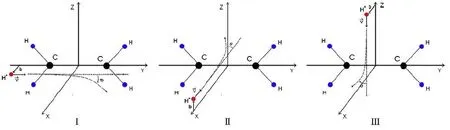
图20.以质子与乙烯碰撞体系为例,展示了三种不同的碰撞方向。黑色、蓝色和红色分别表示碳、氢原子及质子。图片来自[176]
碰撞方向如图20所示,分三种情况:I.在x-y平面,质子初始时刻位于(b,-30a0,0),沿平行于C-C键方向入射;II.在x-z平面,质子初始时刻位于(30a0,0,b),沿x方向垂直于C-C键入射;III.在x-z平面,质子初始时刻位于(b,0,30a0),沿z方向垂直于C-C键入射。碰撞参数b在0~5a0内,步长为0.1a0,在 5.0~10.0a0时,步长为0.5a0。30 eV质子对应的速度为1.43a0/fs,靶体系初始时刻静止。动力学演化时间为40 fs,之间步长为6.05×10-4fs,特殊反应的演化时间会更长。
在碰撞过程中,靶体系的离化机制有两种:直接电子发射(direct electron,即DEE)和电子俘获(EC)。因此,有必要弄清楚 30 eV碰撞能量下靶体系的离化机制。这里以H++C2H2碰撞为例,在其II方向上,碰撞参数为b=0.8 a0。图21展示了沿y和z方向的积分密度ρ(x)随时间的演化过程。质子在14.31 fs时刻进入计算盒子,16.12 fs时,质子周围有一个凸起的密度峰,这是质子的Coulomb场俘获的电子密度。随着时间的演化(17.93 fs),这个密度峰跟随着质子移动。在21.55 fs时,质子进入C2H4分子的电荷密度中心区域,密度峰消失了。这是因为质子俘获的电子数与体系的总电子数相比太小了,以至于密度峰在此时无法识别。质子继续沿着x方向运动,23.36 fs时密度峰又出现了,事实证明,质子碰撞过程中始终能俘获一些电子(见25.17 fs和26.98 fs)。当质子进入吸收边界时(28.79 fs),俘获的电子逐渐被吸收。在30.60 fs时,密度峰越来越小,最终消失不见。对x方向积分可得,整个过程俘获的电子数是0.45,分数电子数可从统计的角度来理解,0.45个电子表示的是质子在多次类似碰撞过程中俘获电子的平均数。假如考虑100个相同的碰撞过程,0.45个总电子数意味着最终将会有45个电子被俘获。在图中总能在质子周围观察到一个小的密度峰,可以认为电子俘获是30 eV下靶离化的基本机制。
除了电子俘获外,碰撞过程中还能够观察到离子间的振动,图22展示了与上图相同事件的原子之间的相对距离。在20 fs之前,所有的键长都处于平衡态,碳原子和质子的距离随着时间增加而单调减小。当质子距离靶目标最近时(20.8 fs),C-C和C-H键中的振动被激发。由于对称性,键长曲线C1-H1(H2)和C2-H3(H4)是重合的。由碰撞诱导的键长变大的现象叫“键稀释”效应,即“bond dilution”effect,该效应最初是在10 eV H++H2的模拟中被发现的,它是由电子暂时从分子轨道移出引起的。实验上和其他理论计算中也发现了类似的“键稀释”效应。本研究中,在C2H2和C2H6中也观察到了“键稀释”效应。在20.8 fs后,碳原子和质子的距离随时间单调递增,这说明了质子被散射而离开体系,整个过程是一个电子俘获散射。

图21.质子与乙烯在方向II上碰撞,b=0.8a0时,电子密度在14.31 fs到30.60 fs的演化。红色小球表示弹离子质子,纵坐标采用对数坐标。蓝色虚线表示边界。图片来自[176]
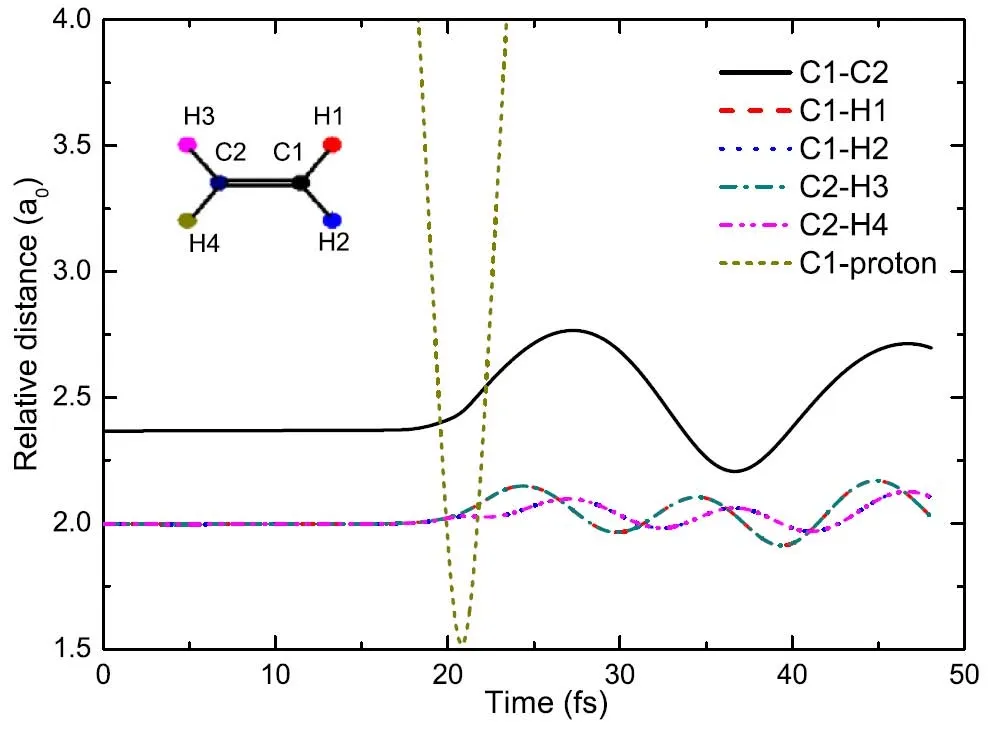
图22.质子与乙烯在方向II上碰撞,b=0.8a0时,原子间的相对距离。图片来自[176]
为了对碰撞过程更加直观的认识,将整个过程可视化,图23显示了H++C2H4在方向I上,且b=1.2 a0时的碰撞过程。质子与靶分子距离很远时,靶体系几乎未受到任何影响 (15.73 fs)。当质子靠近靶体系时(18.15、19.36和20.57 fs),电子密度受到影响,一些电子暂时被质子俘获。当质子离开靶体系时(22.99 fs),它携带了些电子。最终质子会俘获部分电子离开碰撞体系(25.41 fs)。显然,这个过程是电子俘获碰撞,与这与上面的靶离化机制的讨论相符。
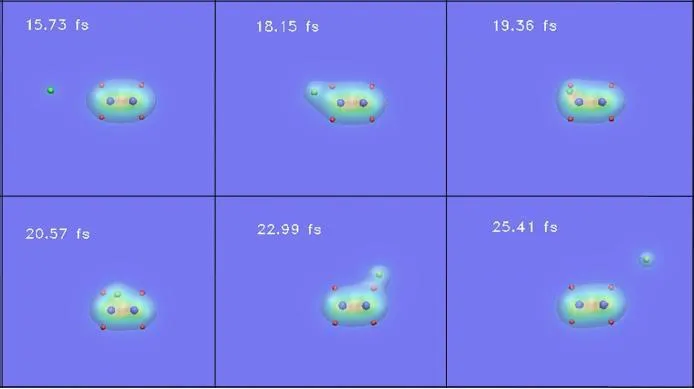
图23.30 eV下,TDLDA-MD模拟在对C2H4碰撞,方向为I,b=1.2 a0。蓝色、红色和绿色小球分别代表碳、氢原子和质子。电子密度从红色到蓝色逐渐减小。图片来自[176]
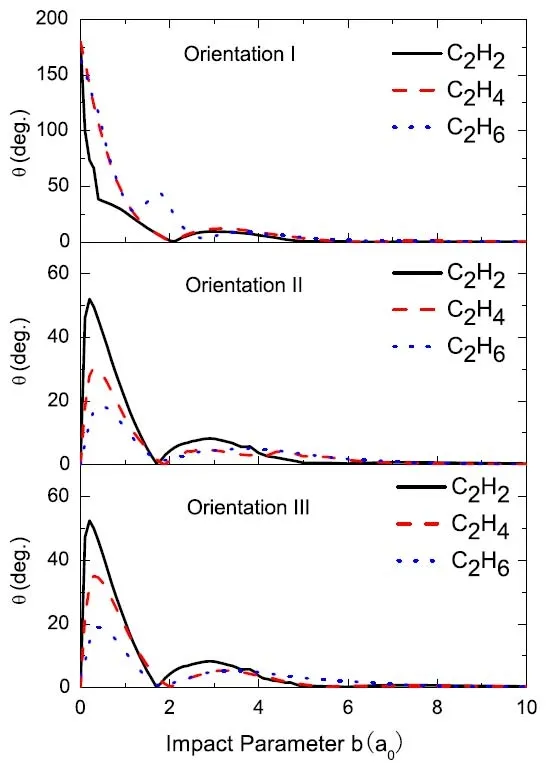
图24.散射角θ与碰撞参数b的函数。图片来自[176]
图24.三个方向上散射角关于碰撞参数b的函数。从力的角度分析了散射角的起源,结果表明,“glory”角的出现是由于动量的净作用效果为零,而不是Giese等人提出的力平衡的过程。实验中,探测到30 eV质子碰撞 C2H2的 “rainbow”峰在3度到8度范围内,本研究计算的 “rainbow”峰,在方向II和 III上是 8.3度,方向I上是9.5度,这与实验值不符合。原因是由方向效应引起的,增加更多的碰撞方向期望可以缩小理论与实验的差距。通过对比其他理论计算的“rainbow”峰值。虽然实验上在30 eV质子与C2H2的碰撞中,由于信噪比太小,没有观察到“rainbow”峰,但是本文给出了“rainbow”峰值,方向I为12.2度和方向II5.4度。另外,没有实验或理论计算涉及到30 eV质子与C2H6的碰撞。
V.结论与展望
本文系统地介绍了五种常见的非绝热动力学方法:经典Ehrenfest方法、面跳跃方法、平均场-面跳跃混合方法(连续面跳跃方法)、混合量子-经典刘维尔方法和从头计算多次产生方法。重点阐述了各种方法的基本思想和数值实现过程,并对其优缺点进行了简要评价。针对不同的研究体系,应根据两点来选择非绝热耦合的处理方法:精确度与计算量,精度越高,计算量越大。
探索新的更有效的非绝热动力学方法任然是今后理论物理与理论化学发展的一个重要课题。理论上可以大致将非绝热动力学划分为两部分:处理非绝热和解决多电子态结构。只要这两部分都用恰当的计算方法处理,解决任何一个精度要求的非绝热过程都不成问题。当然要研究非绝热多自由度复杂体系的前提仍然是寻找到一种数值计算上最简单最有效的方法。因此,在处理非绝热方面,混合量子-经典的方法仍将处于主导地位。而对于多电子态结构方面,密度泛函理论将展露头角,特别是处理激发态的问题。尽管当前用密度泛函来处理激发态的方法很有限,但是对这一领域的研究仍然具有很高的活跃度和挖掘潜力。一旦大量基于密度泛函的方法有成效之后,非绝热分子动力学的可适用范围将被大幅度扩展。正如梯度修正基态密度泛函理论的成功造就了第一性原理(或从头计算)分子动力学的成功和广泛应用。
致谢
感谢国家自然科学基金资助项目(11025524, 11161130520),国家重点基础研究发展计划项目(973计划)(2010CB832903)和欧洲委员会第七框架计划(FP7-PEOPLE-2010-IRSES,269131)项目支持。
参考文献
[1]Born M,Oppenheimer R.Ann Phys Leipzig,1927, 84(20):457-484
[2]Waschewsky G C G,Kash P W,Myers T L,Kitchen D C,Butler L J.J Chem Soc Faraday Trans,1994, 90(12):1581-1598
[3]Reid P J,Lawless M K,Wickham S D,Mathies R A.J Phys Chem,1994,98(22):5597-5606
[4]Jortner J,Levine R D,Rice S A.Adv Chem Phys,1988, 70:1-34
[5]Felker P M,Zewail A H.Adv Chem Phys1988,70:265-364
[6]King D L,Setser D W.Annu Rev Phys Chem,1976, 27:407-442
[7]Kleyn A W,Los J,Gislason E A.Phys Rep,1982, 90(1):1-71
[8]Sayler A M,Leonard M,Carnes K D,Cabrera-Trujillo R,Esry B D,Ben-Itzhak I.J Phys B,2006,39:1701
[9]Luna H,Montenegro E C.Phys Rev Lett,2005,94:043201
[10]Kreibig U,Vollmer M.Optical properties of Metal Clusters.Springer Series in Materials Science:Berlin, 1993
[11]Faisal.TheoryofMultiphoton.Processes.Plenum Press:New York,1987
[12]Zweiback J,Smith R A,Cowan T E,Hays G,Wharton K B,Yanovsky V P,Ditmire T.Phys Rev Lett,2000, 84:2634
[13]Corkum P B,Krausz F.Nat Phys,2007,3:381
[14]Agostini P,DiMauro L F.Rep Prog Phys,2004,67:813
[15]Klessinger M,Michl J.Excited States and Photochemistry of Organic Molecules,VCH.New York,1995
[16]Garavelli M,et al.Faraday Disc,1998,110:51-70
[17]Child M S.Molecular Collision Theory,Academic. New York,1974
[18]Butler L J.Annu Rev Phys Chem,1998,49:125-171 [19]Shuler K E.J Chem Phys,1953,21(4):624-632
[20]Donovan R J,Husain D.Chem Rev,1970,70(4):489-516
[21]Che L,et al.Science.2007,317(5841):1061-1064
[22]Mott N F.Proc Cambridge Philos Soc,1931,27:553-560
[23]Ehrenfest P.Z Phys,1927,45:455
[24]Nikitin E E.Chemische Elementarprozesse,Springer. Berlin,1968
[25]Tully J C,Preston R K.J Chem Phys,1971,55(2):562-572
[26]Tully J C.J Chem Phys,1990,93(2):1061-1071
[27]Drukker K.J Comput Phys,1999,153(2):225-272
[28]Zhang Yan,Xie T X,Han K L.J Phys Chem A,2003, 107:10893-10896
[29]Chua T S.Zhang Y,Han K L.Int Rev Phys Chem, 2006,25:201-235
[30]Gao A H,Li B,Zhang P Y,Han K L.J Chem Phys, 2012,137:204305
[31]Li B,Han K L.J Phys Chem A,2009,113:10189-10195
[32]Li B,Han K L.J Chem Phys,2008,128:114116
[33]Salem L.Electrons in Chemical Reactions:First Principles,Wiley.New York,1982
[34]Salem L,Leforestier C,Segal G,Wetmore R.J Am Chem Soc,1975,97(3):479-487
[36]Massey H S W.Rep Progr Phys,1949,12:248
[37]Stock G,Thoss M.Adv Chem Phys,2005,131:243-375
[38]Tully J C.Classical and Quantum Dynamics in Condensed Phase Simulations,World Scientific.Singapore, 1998
[39]Tully J C.Modern Methods for Multidimensional Dynamics Computations in Chemistry,World Scientific. Singapore,1998
[40]Thorson W,Delos J,Boorstein S.Phys Rev A,1971, 4(3):1052-1066
[41]Micha D A.J Chem Phys,1983,78(12):7138-7145
[42]Gerber R B,Buch V,Ratner M A.J Chem Phys,1982, 77(6):3022-3030
[43]Alimi R,Garc´ıa-Vel A,Gerber R B.J Chem Phys, 1992,96(3):2034-2038
[44]Stock G.J Chem Phys,1995,103(4):1561-1573
[45]Stock G.J Chem Phys,1995,103(8):2888-2902
[46]KosloffR,Hammerich A.Faraday Discuss,1991,91:239-247
[47]Fang J Y,Martens C C.J Chem Phys,1996,104(10):3684-3691
[48]Anderson A.Phys Rev Lett,1995,74(5):621-625
[49]Tully J C,Gomez M,Head-Gordon M.J Vac Sci Technol,1993,11(4):1914-1920
[50]Klein S,Bearpark M J,Smith B R,Robb M A,Olivucci M,Bernardi F.Chem Phys lett,1998,292(3):259-266
[51]Doltsinis N L,Marx D.J Theor Comput Chem,2002, 1:319
[52]Landau L D,Sowjetunion Z.U R S S,1932,2:46
[53]Zener C.Proc R Soc A,1932,137(833):696-702
[55]Miller W H,George T F.J Chem Phys,1972,56(11):5637-5652
[56]Nikitin E E.Theory of Elementary Atomic and Molecular Processes in Gases,Clarendon.Oxford,1974
[57]Herman M F.J Chem Phys,1984,81(2):764-774
[58]Webster F,Rossky P J,Friesner R A.Comput Phys Commun,1991,63(1-3):494-522
[59]Littlejohn R G,Flynn W G.Phys Rev Lett.1991, 66(22):2839-2842
[60]Coker D F,Xiao L.J Chem Phys,1995,102(1):496-510
[61]Bolte J,Keppler S.Phys Rev Lett,1998,81(10):1987-1991
[62]Zhu C,Nakamura H.J Chem Phys,1997,106(7):2599-2611
[63]Zhu C,Teranishi Y,Nakamura H.Adv Chem Phys2001,117:127-233
[64]Nielsen S,Kapral R,Cicotti G.J Chem Phys,2000, 112(15):6543-6553
[65]Horenko I,Weiser M,Schmidt B,Schtte C.J Chem Phys,2002,117(24):11075-11088
[66]Wan C C,Schofield J.J Chem Phys,2000,112(10):4447-4459
[67]Hammes-Schiffer S,Tully J C.J Chem Phys,1994, 101(6):4657-4667
[68]Volobuev Y L,Hack M D,Topaler M S,Truhlar D G.J Chem Phys,2000,112(22):9716-9726
[69]Hack M D,Truhlar D G.J Phys Chem A,2000, 104(34):7917-7926
[70]Blais N C,Truhlar D,Mead C A.J Chem Phys,1988, 89(10):6204-6208
[71]Kuntz P.J Chem Phys,1991,95(1):141-155
[72]Prezhdo O,Rossky P J.J Chem Phys,1997,107(3):825-834
[73]Sholl D,Tully J.J Chem Phys,1998,109(18):7702-7710
[74]Jasper A W,Stechmann S N,Truhlar D G.J Chem Phys,2002,116(13):5424-5431
[75]Ferretti A,Grannucci G,Lami A,Persico M,Villani G.J Chem Phys,1996,104(14):5517-5527
[76]Fang J Y,Hammes-Schiffer S.J Phys Chem A,1999, 103(47):9399-9407
[78]Krylov A I,Gerber R B.J Chem Phys,1996,105(11):4626-4635
[79]Batista V,Coker D.J Chem Phys,1997,106(17): 6923-6941
[80]Chapman S.Adv Chem Phys,1992,82:423-483
[81]Tully J C.Faraday Discuss,1998,110:407-419
[82]P.Pechukas.Phys Rev,1969,181:166-174
[83]Hammes-Schiffer S,Tully J C.J Chem Phys,1995, 103(19):8528-8537
[84]Webster F J,Schnitker J,Friedrichs M S,Friesner R A,Rossky P J.Phys Rev Lett,1991,66(24):3172-3175
[85]Bittner E R,Rossky P J.J Chem Phys,1995,103(18):8130-8143
[86]Bittner E R,Rossky P J.J Chem Phys,1997,107(20):8611-8618
[87]Hack M D,Truhlar D G.J Chem Phys,2001,114(7):2894-2902
[88]Hack M D,Truhlar D G.J Chem Phys,2001,114(21):9305-9314
[89]Domcke W,Stock G.Adv Chem Phys,1997,100:1-169
[90]Manthe U,K¨oppel H.J Chem Phys,1990,93(3):1658-1669
[91]Seidner L,Domcke W.Chem Phys,1994,186(1):27-40
[92]Meyer H D,Miller W H.J Chem Phys,1979,70(7):3214-3223
[93]Topaler M S,Allison T C,Schwenke D W,Truhlar D G.J Chem Phys,1998,109(9):3321-3345
[94]Topaler M S,Allison T C,Schwenke D W,Truhlar D G.J Chem Phys,1999,110(1):687-688
[95]Durup J.Chem Phys,1988,119(1):15-24
[96]Ikegami T,Kondow T,Iwata S.J Chem Phys,1993, 99(5):3588-3596
[97]Truhlar D G,Muckerman J T.Atom-Molecule Collision Theory,Plenum.New York,1979
[98]Kuntz P J.J Chem Phys,1991,95(1):141-155
[99]Prezhdo O V,Rossky P J.J Chem Phys,1997,107(3):825-834
[100]Neumann J V.Mathematical Foundations of Quantum Mechanics,Princeton University.Princeton,1955
[101]Fano U.Rev Mod Phys,1957,29(1):74-93
[102]Mukamel S.Principles of Nonlinear Optical Spectroscopy,Oxford University,New York,1995
[103]Schi ffL I.Quantum Mechanics,McGrow-Hill,1968
[104]Wigner E.Phys Rev,1932,40(5):749-759
[105]Moyal J E.Proc Cambridge Philos Soc,1949,45(1):99-124
[106]Imre K,Ozizmir E,Rosenbaum M,Zweifel P F.J Math Phys,1967,8(5):1097-1108
[107]Hillery M,O’connell R F,Scully M O,Wigner E P.Phys Rep,1984,106(3):121-167
[108]Weyl H.Z Phys,1927,46(1-2):1-46
[109]Schleich W P.Quantum Optics in Phase Space,Wiley-VCH.Berlin,2001
[110]Prigogine I.Non-Equilibrium Statistical Mechanics, Wiley.New York,1962
[111]Jaffe C,Brumer P.J Phys Chem,1984,88(21):4829-4839
[112]Aleksandrov I V.Z Naturforsch,1981,36(8):902-908
[113]Boucher W,Traschen J.Phys Rev D,1988,37(12):3522-3532
[114]Prezhdo O V,Kisil V V.Phys Rev A,1997,56(1):162-175
[115]Martens C C,Fang J.J Chem Phys,1997,106(12):4918-4930
[116]Donoso A,Martens C C.J Phys Chem A,1998, 102(23):4291-4300
[117]Caro J,Salcedo L L.Phys Rev A,1999,60(2):842-852
[118]Kapral R,Cicotti G.J Chem Phys,1999,110(18):8919-8929
[119]Sergi A,Kapral R.J Chem Phys,2003,118(19):8566-8575
[120]Sergi A,Kernan D M,Ciccotti G,Kapral R,Theor Chem Acc 2003,110(2):49-58
[121]Horenko I,Weiser M,Schmidt B,Schtte C.J Chem Phys,2004,120(19):8913-8923
[122]Wan C C,Schofield J.J Chem Phys,2002,116(2):494-506
[123]Santer M,Manthe U,Stock G.J Chem Phys,2001, 114(5):2001-2012
[124]Ando K,Santer M.J Chem Phys,2003,118(23):10399-10406
[125]Shi Q,Geva E.J Chem Phys,2004,121(8):3393-3404
[126]Mak C H,Egger R.Adv Chem Phys,1996,93:39-76
[127]Leforestier C.J Chem Phys,1978,68(10):4406-4410
[128]Car R,Parrinello M.Phys Rev Lett,1985,55(22):2471-2474
[129]Parrinello M.Solid State Comm,1997,102(2-3):107-120
[130]Remler D K,Madden P A.Mol Phys,1990,70(6):921-966
[131]Tuckerman M E,Ungar P J,Vonrosenvinge T,Klein M L.J Phys Chem,1996,100(31):12878-12887
[132]Payne M C,Teter M P,Allan D C,Arias T A, Joannopoulos J D.Rev Mod Phys,1992,64(4):1045-1097
[133]Fantucci P,Bonacic-Koutecky V,Jellinek J,Wiechert M,Harrison R J,Guest M F.Chem Phys lett,1996, 250(1):47-58
[134]Gordon M S,Chaban G,Taketsugu T.J Phys Chem, 1996,100(28):11512-11525
[135]Bolton K,Schlegel H B,Hase W L,Song K Y.Phys Chem Chem Phys,1999,1(6):999-1011
[136]Stich I,Gale J D,Terakura K,Payne M C.Chem Phys Lett,1998,283(5-6):402-408
[137]Hartke B,Carter E A.Chem Phys Lett,1992,189(4-5):358-362
[138]Hartke B,Carter E A.J Chem Phys,1992,97(9):6569-6578
[139]Liu Z,Carter L E,Carter E A.J Phys Chem,1995, 99(13):4355-4359
[140]Hammes-Schiffer S,Andersen H C.J Chem Phys,1993, 99(1):523-532
[141]Maluendes S A,Dupuis M.Int J Quant Chem,1992, 42((5):1327-1338
[142]Jellinek J,Bonacic-Koutecky V,Fantucci P,Wiechert M.J Chem Phys,1994,101(11):10092-10100
[143]Martinez T J,Ben-Nun M,Levine R D.J Phys Chem, 1996,100(19):7884-7895
[144]Ben-Nun M,Quenneville J,Martinez T J.J Phys Chem,2000,104(22):5161-5175
[145]Ben-Nun M,Martinez T J.J Chem Phys,1998, 108(17):7244-7257
[146]Ben-Nun M,Levine R D,Chem Phys,1995,201(1):163-187
[147]Heller E J.J Chem Phys,1981,75(6):2923-2931
[148]Heller E J.J Chem Phys,1975,62(4):1544-1555
[149]王锋.博士论文,(中国科学院,北京,2001)
[150]Wang F,Jiang L,Hong X H,Jiao Y L,Wang J G, Zhang F S.J Chem Phys,2013,139:094108
[151]Wang F,Zhang F S,Abe Y.Chem Phys Lett,2000, 326:461
[152]张艳萍.博士论文,(中国科学院,北京,2007)
[153]Ying M J,Cheng W,Zhang P,Zhang F S.Comp Mat Science,2012,53(1):382
[154]魏宝仁,张丰收,马新文,王锋.原子核物理评论,2002, 19(2):157-160
[155]张丰收,王锋,朱志远,肖国青.物理学报,2001,50(4):667-673
[156]张艳萍,张丰收,蒙克来,肖国青.物理学报,2007,56(4):2092-2097
[157]Mao F,Zhang C,Dai J X,Zhang F S.Phys Rev A, 2014,89:022707
[158]Cheng W,Liu G Q,Zhang F S,Zhou H Y.Phys Lett A,2012,376(45):3363
[159]Cheng W,Ying M J,Zhang F S,Zhou H Y,Ren S F.Nucl Instrum Meth B,2011,269:2067-2074
[160]Mao F,Zhang C,Gao C Z,Dai J X,Zhang F S.J Phys Condens Mat,2014,26:085402
[161]Mao F,Zhang C,Zhang Y W,Zhang F S.Chin Phys Lett,2014,29:076101
[162]Zhang C,Mao F,Zhang F S.J Phys Condens Mat, 2013,25:235402
[163]Zhang C,Mao F,Zhang F S.Eur Phys J Appl Phys, 2013,64:10401
[164]Zhang C,Mao F,Zhang F S,Zhang Y W.Chem Phys Lett,2012,541(5):92
[165]Saalmann U,Schmidt R.Z Phys D,1996,38(2):153-163
[166]Saalmann U,Schmidt R.Phys Rev Lett,1998,80(15):3213-3216
[167]http://pw-teleman.org/
[168]Calvayrac F,Reinhard P G,Suraud E.J Phys B,1998, 31(22):5023-5030
[169]Calvayrac F,Reinhard P G,Suraud E.Ullrich C A.Phys Rep,2000,337(6):493-578
[170]Wang Z P,Dinh P M,Reinhard P G,Suraud E,Zhang F S.Int J Quantum Chem,2011,111(2):480-486
[171]Wang J,Gao C Z,Calvayrac F,Zhang F S.J.Chem. Phys.2014,140:124306
[172]Wang J,Gao C Z,Zhang F S.Chem Phys Lett,2013, 556:256-259
[173]Wang J,Gao C Z,Zhang F S.Nucl Instrum Meth B, 2013,307:277-280
[174]Wang Z P,Wang J,Zhang F S.Chin Phys Lett,2010, 27(5):053401
[175]Wang Z P,Wang J,Zhang F S.Chin Phys Lett,2010, 27(1):013101
[176]Gao C Z,Wang J,Zhang F S.Chem Phys,2013,410:9-18
[177]Gao C Z,Wang J,Zhang F S.Nucl Instrum Meth B, 2013,307:225-228
[178]Gao C Z,Wang J,Zhang F S.J Chem Phys.,2014,140:054308
[179]Heidrich D,van Eikema Hommes N J R,von Ragu′e Schleyer P.J Comput Chem,1993,14:1149
[180]Marx D,Parrinello M.Science,1999,284:59
[181]Kamarchik E,Mazziotti D A.Phys Rev A,2009,79:012502
The application of Born-Oppenheimer approximation has received a great success,however it fails to treat dynamical process involving nonadiabatic transition.Therefore,the description of ionic motion coupled with electrons must be taken into account.The nonadiabatic process is extremely common in the field of photophysics,photochemistry,strong-field physics and ion collisions.Electron excitation is essential in nonadiabatic process,which is closely connected with ground state and excited state.Basically full quantum mechanical methods can deal with nonadiabatic electron-ion coupling without making any approximations,but it is presently restricted to small systems consisting of two or three atoms.The computational cost increases exponentially with increasing the number of atom when to exactly solve quantum mechanics equations,which is far too difficult to treat nonadiabatic process in atomic system.Over recent decades,several groups have developed many different methods to study the nonadiabatic process.This work mainly introduces the nonadiabatic mixed quantum-classical method,where nuclei are treated with classical limit,and electrons are described by quantum theory.Mixed quantum-classical method includes Ehrenfest method,Surface hopping method,Hybrid method(combined merits of Ehrenfest method with Surface hopping method),and Liouville method based on Wigner transform.However,when the quantum effects of nuclei are remarkable,nonadiabatic dynamics must resort to other more rigorous method,such as ab initio multiple spawning.This article briefly and clearly introduces relevant concepts,numerical implementation,recent progress and its pros and cons.
Progress in nonadiabatic coupled electon-ion dynamics
Wan Shi-Zheng1,2,Wang Jing1,2,Gao Cong-Zhang1,2,Zhang Feng-Shou1,2,3
1.The Key Laboratory of Beam Technology and Material Modification of Ministry of Education, College of Nuclear Science and Technology,Beijing Normal University,Beijing 100875,China
2.Beijing Radiation Center,Beijing 100875,China
3.Center of Theoretical Nuclear Physics,National Laboratory of Heavy Ion Accelerator of Lanzhou, Lanzhou 730000,China
Nonadiabatic;Coupling mixed quantum-classical method;Molecular dynamics; Excited state
O571.25+1
A
2014-6-30
∗ fszhang@bnu.edu.cn
1000-0542(2014)05-0203-23 203