金磁纳米微粒表面蛋白偶联率的测定
2014-06-27马建中崔亚丽
杨 冬, 马建中, 高 敏, 崔亚丽
(1.陕西科技大学 化学与化工学院, 陕西 西安 710021; 2.陕西科技大学 资源与环境学院, 陕西 西安 710021; 3.西北大学 生命科学学院, 陕西 西安 710069)
0 引言
金磁纳米微粒是表面包覆金壳的磁性纳米复合微粒,因其兼具胶体金独特的光学性质及磁性材料的超顺磁性,而在生物分离[1]、免疫学检测[2]、靶向给药[3]、基因转导[4]、固定化酶[5]等诸多生物医学领域中有着广泛的应用前景.
当将金磁纳米微粒与生物分子,特别是与免疫活性物质(抗原或抗体)结合并用以免疫检测时,蛋白分子在颗粒表面的有效偶联是构建免疫探针及实施后续检测的前提.其中,偶联率的确定可以为分析蛋白分子在颗粒表面的空间分布提供有效依据,且对检测效果,主要指特异性及灵敏度的评价分析具有现实意义[6-8].
基于金磁纳米微粒的表面特性,本实验采用1-(3-二甲氨基丙基)-3-乙基碳二亚胺(1-(3-dimethyllaminopropyl)-3-ethylcarbodiimide hydrochloride,EDC)作为连接因子,以化学键合的方式将链亲合素(Streptavidin,SA)分子偶联于金磁微粒表面,排除定量方法中的干扰因素和物质,以Lowry蛋白定量法测得准确的偶联率,通过金磁纳米微粒的密度的计算,并结合蛋白分子的分子量计算得出链亲合素在颗粒表面的分布个数,为基于金磁纳米微粒的免疫探针的结构分析提供了一种可靠的方法.
1 实验部分
1.1 主要试剂
金磁纳米微粒,十六烷基三甲基溴化铵(CTAB),聚丙烯酸钠(PAA),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC),链亲合素(SA),三氯乙酸(分析纯),Lowry试剂盒.
1.2 主要仪器
UV-2550型紫外可见分光光度计,日本岛津;VORTEX 1 MS 3型涡旋混匀器,德国IKA;5425D型离心机,德国Eppendorf;5700型傅立叶红外光谱仪,美国Nicolet;电感耦合等离子体原子发射光谱,Thermo Elemental;移液器,德国Eppendorf.
1.3 实验方法
1.3.1 金磁纳米微粒表面修饰及元素分析
取金磁纳米微粒30 mg(5 mg/mL),加入等体积的CTAB溶液(5 mmol/L),搅拌30 min,置于磁性分离器上磁性分离,弃去上清液,以去离子水重悬.加入5 mL聚丙烯酸钠(PAA,1 mg/mL),搅拌60 min,反应结束后弃去上清液,洗涤3次,最后以去离子水定容至10 mL,备用.取1 mL修饰后的金磁纳米微粒于真空烘箱过夜,称重.
将表面修饰的复合粒子稀释成稀溶液,用移液器吸取30μL滴加在碳膜覆盖的铜网上,充分干燥后,在透射电子显微镜(TEM)下观察修饰后的微粒分散度及表面形貌.
取一定量的金磁纳米微粒于玻璃皿中,置于80 ℃的烘箱中干燥过夜.取粉末样品进行红外光谱(FTIR)分析,表征微粒表面特征官能团.
取表面修饰后的微粒10 mg,加入王水溶化,过夜烘干后,以去离子水重悬.采用电感耦合等离子发射仪(ICP-AES)测定元素金及铁的含量,计算金磁纳米颗粒的密度.
1.3.2 Lowry蛋白定量
(1)标准曲线的绘制
Lowry蛋白定量的原理见文献[9],Lowry试剂盒操作步骤如下:
①分别取A2、A3、A1液体,按1∶1∶100顺序混合,放置0.5 h后使用.
②取标准品BSA和SA,分别以去离子水稀释为0μg/mL、10μg/mL、20μg/mL、40μg/mL、80μg/mL、160μg/mL、320μg/mL、640μg/mL、1 280μg/mL的标准品系列溶液.
③分别取标准品系列溶液20μL,加入A液1 mL,放置0.5 h后加入B液100μL,迅速混匀(避光操作),旋转0.5 h,以紫外可见分光光度计测定A750值,每个样品做三次平行,取平均值绘制标准曲线.
(2)EDC对蛋白定量的测定干扰规律
配制样品中蛋白含量为50μg/mL,EDC终浓度分别为0μg/mL、10μg/mL、20μg/mL、40μg/mL、80μg/mL、160μg/mL、320μg/mL、640μg/mL、1 280μg/mL的样品液系列,按前述方法测定样品的蛋白含量,比较加EDC前后蛋白含量的测定值.
(3)对EDC干扰的消除
取样品液1 mL,加0.15 mL三氯乙酸溶液,混匀,室温放置15 min,4 ℃,12 000 rpm离心5 min,小心地吸弃上清,重复上述步骤2次,Lowry法测定蛋白含量.
1.3.3 金磁纳米微粒偶联蛋白及偶联率的计算
取1 mg金磁纳米微粒,磁分离弃去上清液,以0.2 mL磷酸缓冲液(PB×1,pH 6.7)重悬,加入20μg EDC溶液(10 mg/mL),冰浴超声处理20 min,加入SA溶液(5 mg/mL),使其终浓度为50μg/mL,超声处理60 min,磁分洗涤3次,0.2 mL的PB缓冲液4 ℃保存,取浓度50μg/mL的SA溶液、磁分得到上清液,以及第一次的洗涤液,分别测定蛋白含量,计算偶联率.
2 结果与讨论
2.1 金磁纳米微粒的表征
金磁纳米微粒的表面修饰原理参见文献[10].从图1(a)可以看出,采用CTAB及PAA对微粒表面修饰后,可以得到颜色红亮的微粒溶液.由于金磁纳米微粒的表面等离子体共振现象,金磁纳米微粒在可见光范围内存在一个吸收光谱带,其最大吸收峰位在540 nm左右,且其位置会随粒径分布、分散介质的变化及材料表面组成等而产生相应的红移或蓝移[11].当金磁纳米微粒溶液呈现红亮的颜色时,代表其具有良好的分散稳定性.表面修饰后的金磁纳米颗粒为规则的球状(如图1(b)所示),通过Nano Measure计算,其平均粒径为41.89 nm.
为确定金磁纳米微粒的表面修饰效果,将修饰后的微粒反复洗涤,去除未包裹的PAA后,80 ℃真空烘箱干燥, 以红外光谱分析表面特征官能团.如图1(c)所示,表面修饰前的金磁微粒的2 920 cm-1, 2 850 cm-1, 1 401 cm-1, 以及 960 cm-1等处的吸收峰来自于表面活性剂CTAB,即2 920 cm-1及2 850 cm-1处的吸收峰来自于C-H的对称伸缩振动,1 401 cm-1为C-H的不对称振动,而960 cm-1处的吸收峰则来自于CTAB的季胺盐极性头[12,13].中性溶液中,PAA链上的羧基会产生部分解离形成羧酸根,因而在1 550 cm-1和1 400 cm-1处出现特征峰[14,15].
对比表面修饰前后的谱图,修饰后的金磁微粒具有来自于聚丙烯酸羧基官能团,表明PAA通过羧基与CTAB的季胺极性头包裹在了颗粒外层,形成的聚合物外壳可以为颗粒提供离子稳定性及空间位阻,从而保持颗粒具有良好的分散稳定性[10,16].
通过电感耦合等离子体原子发射光谱(ICP-AES)的元素定量分析(如图1(d)所示),结果表明表面修饰后的金磁纳米微粒中Au含量为10.8%、Fe含量约为16.2%,即:
mAu∶mFe3O4=10.8∶22.4
如果将金磁纳米微粒看作是均匀的规则球体颗粒,如图1(d)所示[17],则其密度计算公式如下 :
其中,ρAu=19.3×106g/m3,ρFe3O4=5.18×106g/m3,求解出d=14.5 nm,计算出金磁纳米微粒的平均密度为ρFe3O4@Au=18.71×106g/m3,则金磁纳米微粒的浓度N=1.46×1012个/mg.
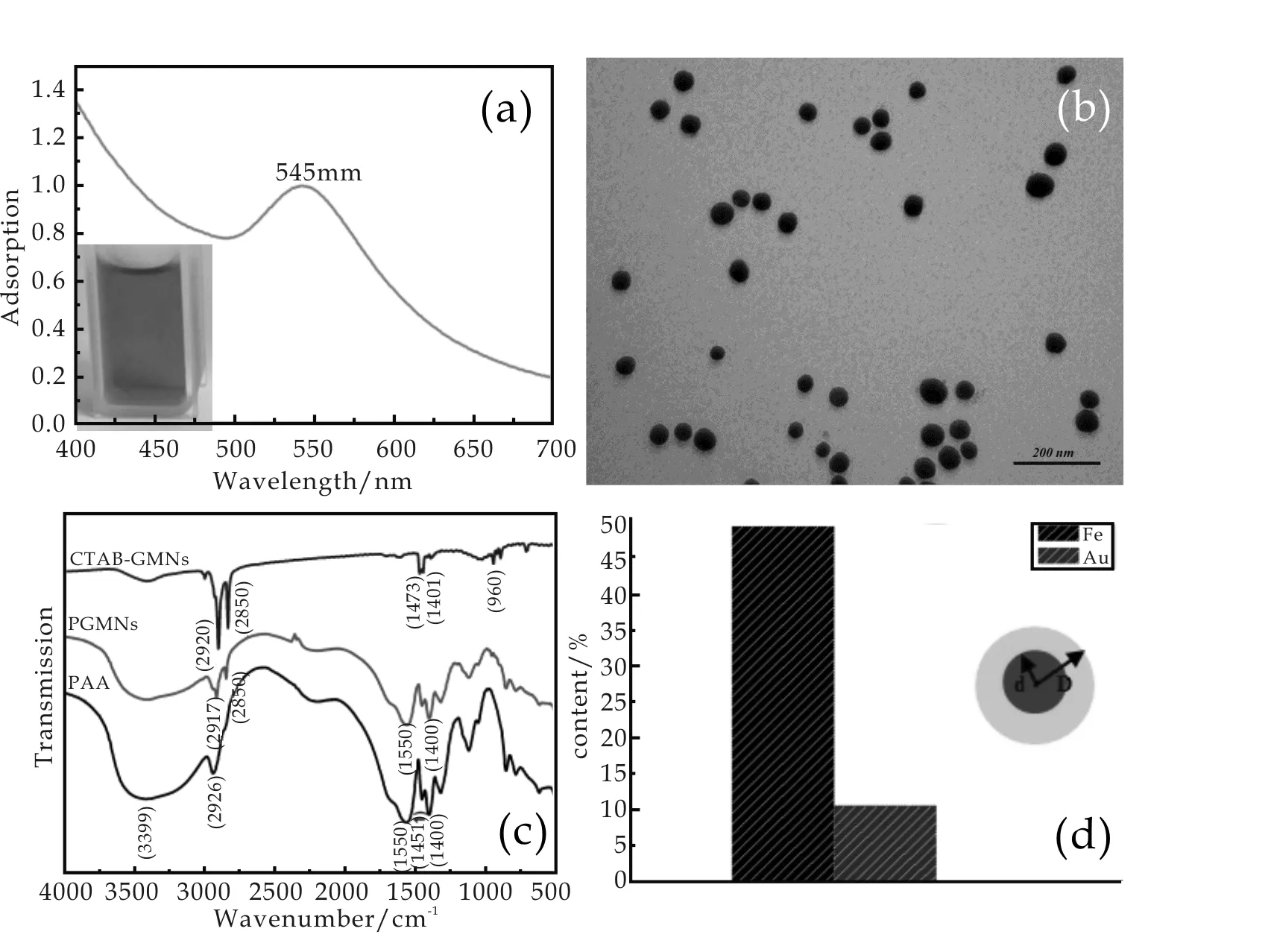
(a)金磁纳米微粒及表面等离子体共振吸收峰 (b)金磁纳米微粒的透射电子显微镜图(TEM) (c)红外光谱谱图(FTIR) (d)电感耦合等离子体原子发射光谱(ICP-AES)表征的元素定量分析图1 金磁纳米微粒的表征结果
2.2 蛋白定量的标准曲线
除了操作过于繁琐的凯氏定氮法,以及灵敏度差的紫外吸收法外,还可以应用较为广泛的蛋白定量方法包括考马斯亮蓝法(Bradford法),二喹啉甲酸法(BCA法)以及Lowry法(Folin-phenol reagent)等.然而,Bradford法形成的化合物的光吸收值在595 nm,BCA法形成的复合物的光吸收值在562 nm,使得金磁纳米微粒本身特征峰位的移动都会对定量结果产生巨大影响,因而,选择Lowry法进行蛋白定量.
以SA和BSA为标准品绘制得到的标准曲线如图2所示.结果表明,蛋白含量-吸光度的线性关系都比较显著,然而两种蛋白得到的标准方程明显存在区别.这是由于蛋白质中含有酪氨酸和色氨酸残基的量的不同,导致了等量的不同蛋白所显示的颜色深度不一致,因而产生了误差[18].因此,本实验以需要偶联的目标蛋白SA作为标准品,标准方程为A=4.165×10-4c+0.011,标准偏差为0.994.
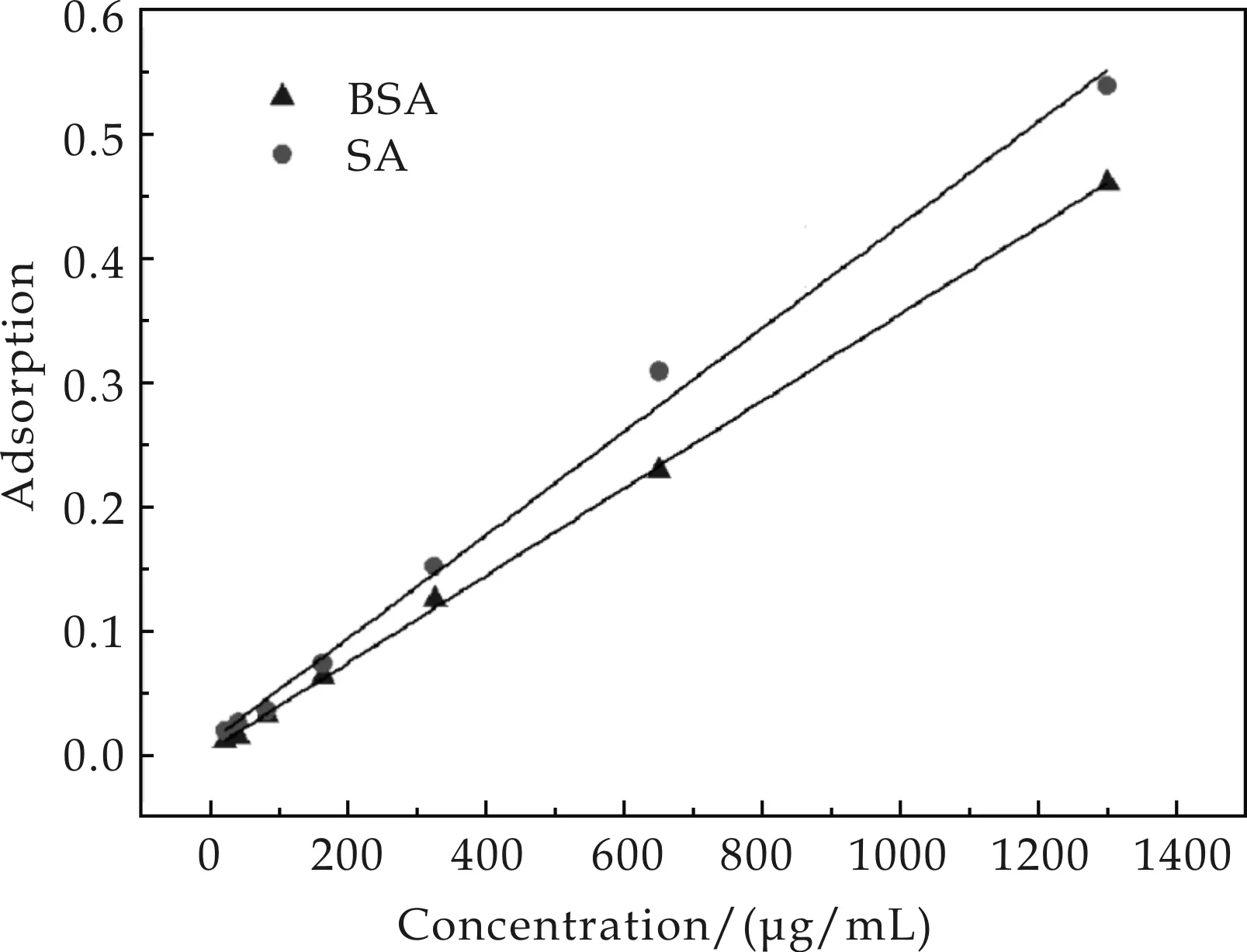
图2 Lowry法中以链亲合素(SA)和牛血清 蛋白(BSA)作为标准品绘制的标准曲线
2.3 EDC对蛋白定量的干扰
在纳米粒表面偶联蛋白,可以通过物理吸附和化学键合的方法.物理吸附对纳米粒的表面特性要求较高,吸附于纳米粒表面的蛋白分子易于脱落而重新进入到溶液中,因而能过物理吸附得到的蛋白标记纳米粒不稳定.为得到更稳定的蛋白功能化金磁微粒,我们采用碳二亚胺(EDC)作为连接因子,通过金磁微粒表面的羧基与肽链上的胺基反应,将蛋白分子化学键合至金磁纳米微粒表面,其反应示意图如图3所示.
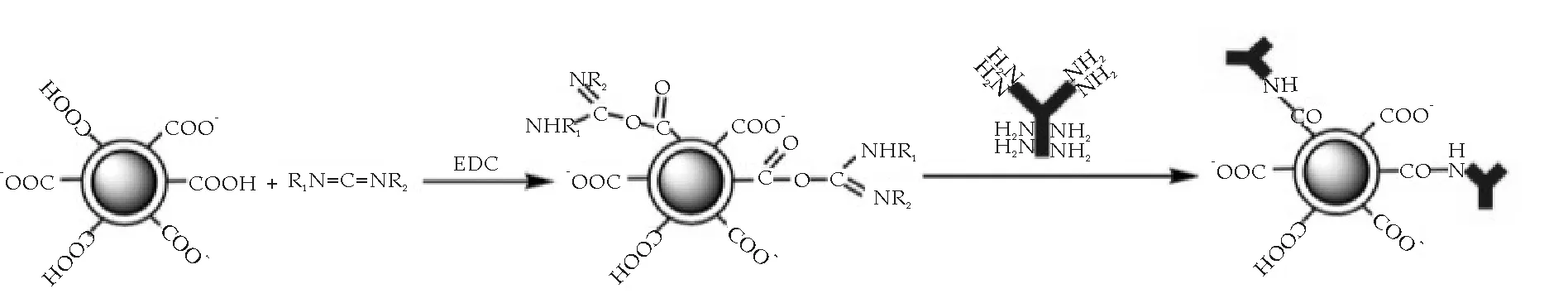
图3 碳二亚胺(EDC)作为连接因子实现 金磁纳米微粒偶联蛋白的示意图
然而,实验中发现微克级的碳二亚胺就会对
Lowry蛋白定量法产生干扰[19,20],如图4所示.以不含SA的系列浓度EDC溶液来进行Lowry法检测.结果表明,EDC浓度-吸光度符合直线关系,标准方程为A=4.421×10-4c+0.012,其标准偏差为0.999.
为研究EDC对蛋白定量的干扰,配制了含有SA终浓度为50μg/mL、EDC终浓度为0μg/mL、10μg/mL、20μg/mL、40μg/mL、80μg/mL、160μg/mL、320μg/mL、640μg/mL、1 280μg/mL的样品液系列.按前述方法测定样品的蛋白含量,对比得到EDC对蛋白定量的影响.结果表明,在有蛋白存在时,小浓度的EDC(c<200μg/mL)对蛋白定量的影响较小,可以通过EDC的线性方程去除干扰,然而随着EDC浓度的增大,所产生的干扰愈加严重,因而难以得到准确的测定值.
在纳米颗粒偶联蛋白的操作过程中,往往需要加入过量的EDC,以提高蛋白在颗粒表面的偶联,得到最大的偶联率.所以,偶联率的测定必须消除体系中EDC对Lowry法的干扰.
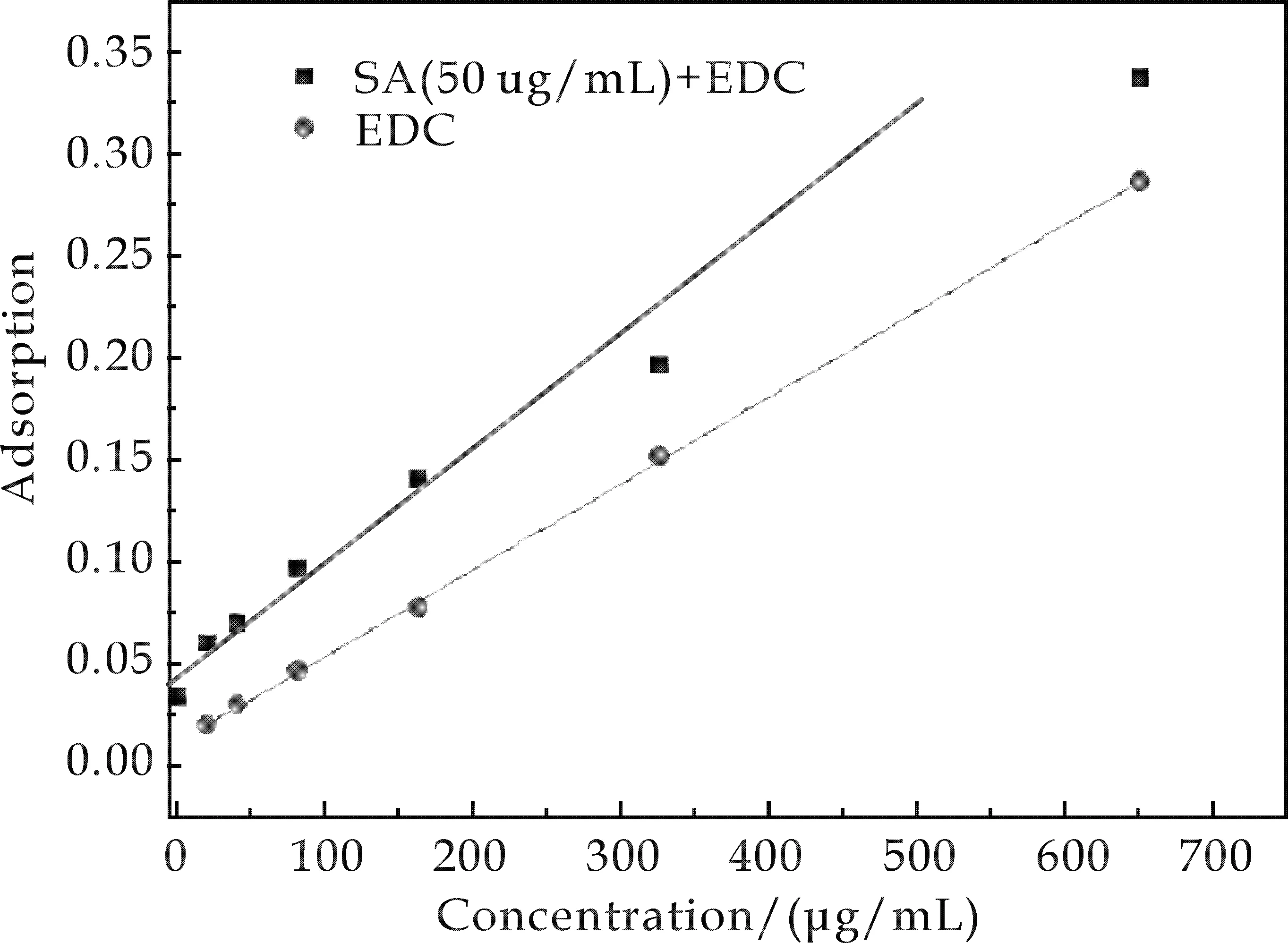
图4 EDC对Lowry蛋白定量法的干扰
2.4 对EDC干扰的消除
采用三氯乙酸沉淀蛋白后,再进行Lowry法蛋白定量所得的结果如表1所示.可以看出,当蛋白中混有不同浓度的EDC时,可参照《欧洲药典》引入三氯乙酸,先沉淀蛋白后进行测定,可以消除EDC引起的干扰现象,从而准确定量体系中的蛋白含量.
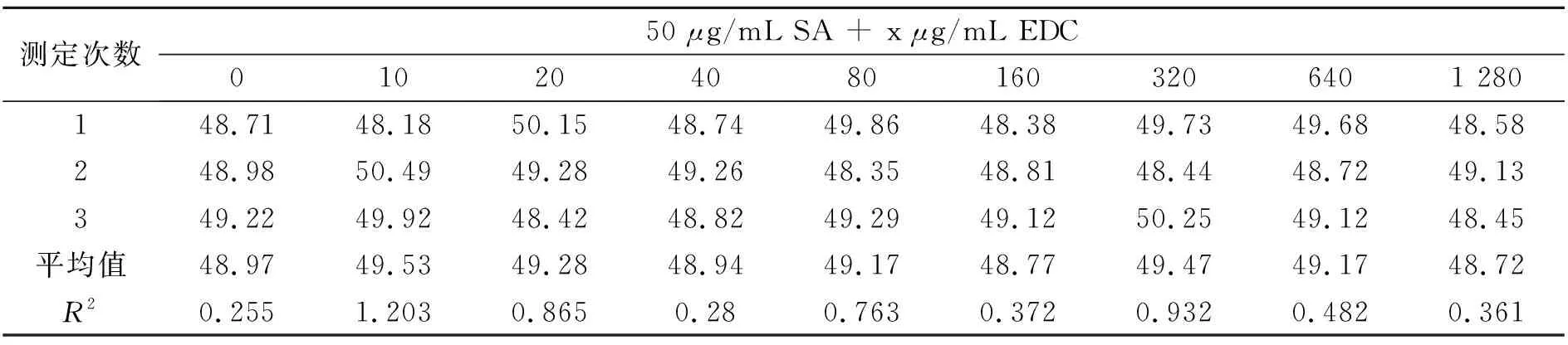
表1 Lowry法检测含50 μg/mL链亲合素(SA)的不同浓度的EDC溶液
2.5 偶联率的评价
金磁纳米微粒表面偶联蛋白前后,取50μg/mL的SA溶液作为Apre样品液,以三氯乙酸沉淀偶联后蛋白作为Apost样品液,并收集洗涤液作为Awash样品液,通过Lowry法进行蛋白定量,测定结果如表2所示.
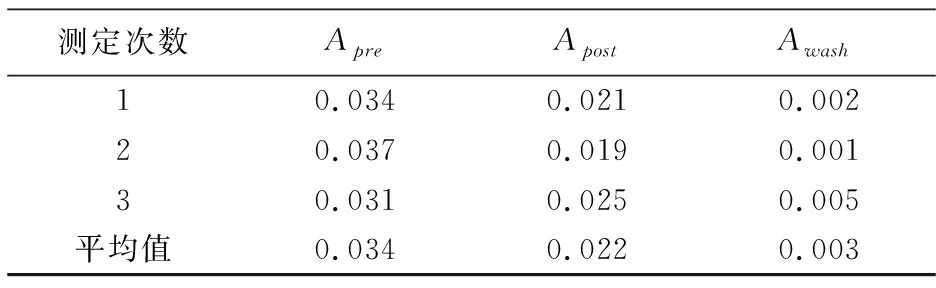
表2 偶联前后蛋白定量结果
SA在溶液中的含量减少表明SA在金磁纳米微粒表面成功偶联.由于EDC作为连接因子,在后续洗涤中SA被洗脱的量很少,其中偶联率的评价公式为:
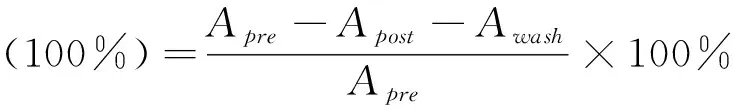
通过计算可得,金磁纳米微粒表面SA的偶联率达到28.43%,结合SA的相对分子量以及金磁钠米微粒的浓度,其偶联量为:
其中,L=6.02×1023,N=1.46×1012个/mg,MSA=64 kDa,则计算所得偶联量为18个SA/个金磁微粒.
3 结论
对纳米微粒进行定量表面功能化,不但可为免疫探针结构的研究提供了空间分布信息,也为后续检测性能的评价提供了参考数据及评价依据.因此,本研究通过微粒表面的蛋白偶联率的测定和计算,为构建基于金磁纳米微粒的免疫探针,提供了一定的理论依据和技术参考.
在本研究的计算方法中,采用了平均粒径来计算金磁纳米微粒的密度.因为纳米级微粒的粒径分布大多符合正态分布,应是具有一定范围的粒径分布[21],因而会导致结果出现一定的偏差;同时,本研究中以三氯乙酸来消除EDC对Lowry蛋白定量的干扰,由于离心去上清等操作的有限性,可能溶液中会有EDC的残留,从而导致偶联后的蛋白定量结果略偏大.因此,本研究的计算结果只能为实验提供一定参考,真正精确的微粒表面的蛋白定量还需要进一步努力.
综上所述,本文以聚电解质PAA修饰金磁纳米微粒,采用EDC将SA分子偶联于微粒表面,用三氯乙酸沉淀法来消除溶液中未反应的EDC对Lowry蛋白定量方法的干扰,确定了微粒表面蛋白的偶联量,同时结合金磁纳米微粒密度及SA分子量的数据,计算得出金磁纳米微粒表面SA的偶联量为18个SA/金磁微粒.
[1]Park H Y,Schadt M J,Wang L,et al.Fabrication of magnetic core @shell Fe oxide @Au nanoparticles for interfacial bioactivity and bio-separation[J].Langmuir,2007,23(17):9 050-9 056.
[2]Liang W,Yi W,Li Y,et al.A novel magnetic Fe3O4@gold composite nanomaterial:Synthesis and application in regeneration-free immunosensor[J].Materials Letters,2010(64):2 616-2 619.
[3]Chao X,Guo L,Zhao Y,et al.PEG-modified Gold Mag nanoparticles (PGMNs) combined with the magnetic field for local drug delivery[J].Journal of Drug Target,2011,19(3):161-170.
[4]Sun H,Zhu X,Zhang L,et al.Capture and release of genomic DNA by PEI modified Fe3O4/Au nanoparticles[J].Materials Science and Engineering:C,2010,30(2):311-315.
[5]王显祥,黄 硕,单 志,等.自组装制备Fe3O4@Au复合纳米粒子用于固定化葡萄糖氧化酶[J].科学通报,2009,54(4):430-435.
[6]Minkstimiene A K,Ramanaviciene A,Kirlyte J,et al.Comparison of oriented and random antibody immobilization techniques on the efficiency of immunosensor[J].Analytical Chemistry,2010, 82:6 401-6 408.
[7]Lin P C,Chen S H,Wang K Y,et al.Fabrication of oriented antibody-conjugated magnetic nanoprobes and their immunoaffinity application[J].Analytical Chemistry,2009,81(21):8 774-8 782.
[8]Moyano D F,Rotello V M.Nano meet biology:structure and function at the nanoparticle interface[J].Langmuir,2011,27(17):10 376-10 385.
[9]Markwell M A K,Haas S M,Bieber L L,et al.A modification of the lowry procedure to simplify protein determination in membrane and lipoproteion samples[J].Analytical Biochemistry,1978,87(1):206-210.
[10]Yang D,Ma J Z,Zhang Q L,et al.Polyelectrolyte coated gold magnetic nanoparticles for immunoassay development: toward point of care diagnositics[J].Analytical Chemistry,2013,85(14):6 688-6 695.
[11]Yang D,Ma J Z,Gao M,et al.Suppression of composite nanoparticle aggregation through steric stabilization and ligand exchange for colorimetricprotein detection[J].RSC Advances,2013,3:9 681-9 686.
[12]Nikoobakht B,El Sayed M A.Evidence for bilayer assembly of cationic surfactant on the surface of gold nanorods[J].Langmuir,2001,17:6 368-6 374.
[13]Wijaya A,Hamad Schifferli K.Ligand customization and DNA functionalization of gold nanorods via round-trip phase transfer ligand exchange[J].Langmuir,2008,24:9 966-9 969.
[14]Choi J,Rubner M F.Influence of the degree of ionization on weak polyelectrolyte multilayer assembly[J].Macromolecules,2004,38:116-124.
[15]Kato N,Schuetz P,Fery A,et al.Thin multilayer films of weak polyelectrolytes on colloid particles[J].Macromolecules,2002,35:9 780-9 787.
[16]Yang D,Ma J Z,Peng M L,et al.Building nanoSPR biosensor systems based on gold magnetic composite nanoparticles[J].Journal of Nanoscience and Nanotechnology,2013,13(8):5 485-5 492.
[17]Yu C,Varghese L,Irudayaraj J.Surface modification of cetyltrimethylammonium bromide-capped gold nanorods to make molecular probes[J].Langmuir,2007,23:9 114-9 119.
[18]Kumar R,Shukla A K,Bagga E,et al.1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide interference with Lowry method[J].Analytical Biochemistry,2005,336:132-134.
[19]张云茹,周跃刚,范守城,等.硫酸铵存在下的改良福林-酚试剂法[J].重庆工学院学报(自然科学版),2008,22(7):159-166.
[20]Winters A L,Minchin F R.Modification of Lowry assay to measure proteins and phenols in covalently bound complexes[J].Analytical Biochemistry,2005,346:43-48.
[21]李 晖,刘 运,张方辉,等.ZnO纳米颗粒的分散及光致发光特性[J].陕西科技大学学报(自然科学版),2013,31(1):29-32.
