石油加氢废催化剂中钨和镍的提取及镍的酸浸动力学
2014-06-04许傲云郑成志赵曙辉
许傲云,郑成志,赵曙辉
(东华大学 化学化工与生物工程学院,上海 201620)
石油加氢废催化剂中钨和镍的提取及镍的酸浸动力学
许傲云,郑成志,赵曙辉
(东华大学 化学化工与生物工程学院,上海 201620)
研究了从石油加氢废催化剂中回收钨、镍的方法,通过正交试验考察了提取钨和镍的最佳工艺条件,并对镍的浸出过程动力学进行了研究.结果表明:当Na2CO3的用量是WO3理论量的6倍,在750℃下钠化焙烧4 h,焙料在90℃下水浸2 h,WO3的浸出率可达到95%以上;镍富集在浸出渣中,在硫酸质量分数为30%,固液比为1∶8,85℃下浸出4 h,催化剂中镍的浸出率可达到98%以上;镍的浸出过程属于扩散控制模型,与扩散控制动力学方程式相吻合,浸出反应的表观活化能为15.95 kJ/mol.
废催化剂;钨;镍;扩散控制模型;酸浸动力学
催化剂被广泛应用于石油炼制与化工行业,现代化工生产中约有80%的反应离不开相应的催化剂.全世界每年消耗的催化剂数量约为80万t(不包括烷基化用的硫酸和氢氟酸催化剂),其中,炼油催化剂约为41.5万t,化工催化剂约为33.5万t,环保用催化剂约为4.7万t[1].催化剂中通常含有铝硅载体,如钼、钴、钒、镍等稀有金属,这些金属具有特殊的结构和活性,可以提高原料的去杂率,例如,金属钒可以极大地提高原料加氢脱硫、脱氮、脱金属的效率.近年来,为了满足日益增长的超低硫燃料的需要,不断提高对催化剂的石油加氢能力,催化剂经过几次循环再生利用,其活性下降或有些催化剂永久性中毒,使其不再具有可再生利用价值,因此,每年有大量的废催化剂排放.但废催化剂中仍然含有相当数量的有色金属或贵金属,有时它们的含量会远远高于贫矿中相应组分的含量[2].废催化剂作为二次资源,如果直接排放既会造成资源的浪费,也会对环境造成极大的污染.开展废催化剂的综合回收,不仅有利于减少环境污染,实现资源的循环利用,而且有利于催化剂生产企业降低生产成本,提高技术服务水平和产品市场竞争力[3-4].
目前,国内外对废催化剂中有用金属的回收已进行了大量的研究与报道,但主要是针对钴-镍体系[5]、钒-钼体系[6]、钴-钼体系[7],对钨的综合回收利用的报道鲜见.本文所研究的废催化剂中含有相当可观的钨,其次是镍,因而具有很大的回收利用价值和经济效益.本文采用钠化焙烧-水浸提取钨和酸浸提取镍工艺,对废催化剂中的钨和镍进行回收,其工艺流程简单,成本低,提取率高,工艺可行性强.国内对镍的酸浸回收大多侧重于工艺方面,对镍的浸出反应的机理研究较少.为了探讨镍的浸出规律,研究浸出反应的控制步骤,本文还对提取镍的酸浸过程动力学进行了研究.
1 试验药品和仪器
1.1 试验药品与所用材料
废催化剂,上海中河有色金属公司,黑色(略带绿色)颗粒状,由IE300X型能谱仪测试其主要化学成分,结果如表1所示.
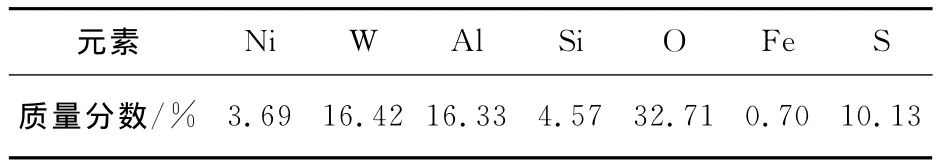
表1 废催化剂的主要成分Table 1 Main ingredients of spent catalyst
试剂:碳酸钠、氢氧化钠、盐酸、硫酸等均为分析纯.
1.2 试验仪器
密封式化验制样粉碎机,Ls13320型激光粒度分析仪,SX-4-10型马弗炉,H2004G型强力电动搅拌器,WZ-100SP型恒温水浴锅,DF-101B型集热式磁力搅拌器,GM-0.33A型隔膜真空泵,722N型可见分光光度计,876-L型真空干燥箱等.
2 试验方法
2.1 氧化焙烧
将废催化剂在真空干燥箱内于100℃下烘48 h,除去其水分,再置于密封式化验制样粉碎机粉碎,用激光粒度分析仪测得粒径为0.175~0.261 mm.将一定质量经研磨筛分后的废催化剂放入马弗炉中,在650℃下氧化焙烧4 h(实际焙烧以催化剂无黑心时,即为焙烧终点),将硫化物转化为氧化物,主要反应为
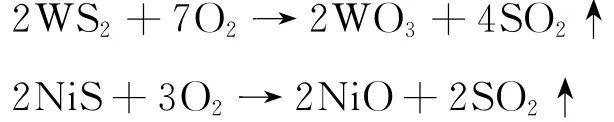
同时除去废催化剂中的水分、有机杂质和积碳物.
2.2 钠化焙烧
将废催化剂与碳酸钠以一定的比例置于马弗炉中高温焙烧,可以将 WO3转化为可溶的钨酸钠,控制氧化钨与碳酸钠浸渍焙烧反应条件,可使95%以上的WO3转化,主要反应为
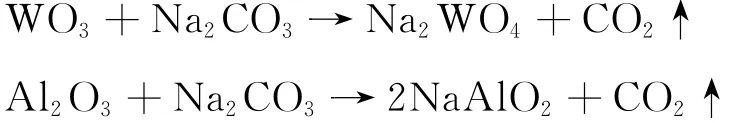
2.3 浸出试验
水浸钨:浸出试验在恒温水浴槽中进行,机械搅拌,水浴加热.浸出完成后,过滤,重复以上操作,将两次的滤液合并,用硫氰酸盐比色法[8]测定浸滤液中WO3的浓度,计算其浸出率.得到的粗钨酸钠溶液经过除杂分离出铝后,加入CaCl2得到钨酸钙沉淀,根据需求再制得不同的钨制品.
酸浸镍:称取一定量提取钨以后的滤渣,倒入250 m L三口烧瓶中,按照一定的液固比往烧瓶中加入稀硫酸,然后将烧瓶放入恒温水浴中搅拌浸出.浸出完成后,过滤,用丁二酮肟分光光度法[9]测定浸滤液中镍的浓度,计算其浸出率.
3 结果与讨论
3.1 钠化焙烧条件的影响因素与分析
以650℃下氧化焙烧4 h后的样品经预备试验和单因素条件试验,确定了影响 WO3浸出率的焙烧因素主要为碳酸钠的用量、钠化焙烧温度和焙烧时间以及各个因素的主要影响范围.为了考察碳酸钠的用量、钠化焙烧温度和焙烧时间相互作用对WO3浸出率的影响,进一步得到钠化焙烧阶段的最优工艺条件,在水浸温度为90℃、水浸时间为2 h、固液比为1∶3的条件下,对焙烧产物进行3因素3水平的L9(33)的正交试验分析,试验结果如表2所示.

表2 钠化焙烧正交试验结果Table 2 Sodium baking orthogonal experiments results
由表2的正交试验结果得到钠化焙烧各因素对WO3的浸出率影响如图1所示.由图1可知,在钠化焙烧影响因素中,焙烧温度的曲线波动较大,对WO3的浸出率影响最大,焙烧时间次之,Na2CO3的用量影响较小.但由图1(a)可以看出,当Na2CO3的用量由WO3理论用量的6倍增加到7倍时,WO3浸出率只提高了0.33%,说明当Na2CO3的用量为WO3理论用量6倍时已足够将钨转化为钨酸钠,继续增加碳酸钠用量,只是增加了工艺成本,而没有明显提高浸出率.因此,最佳各因素水平组合:Na2CO3的用量为WO3理论用量的6倍,焙烧时间为4 h,焙烧温度为750℃.
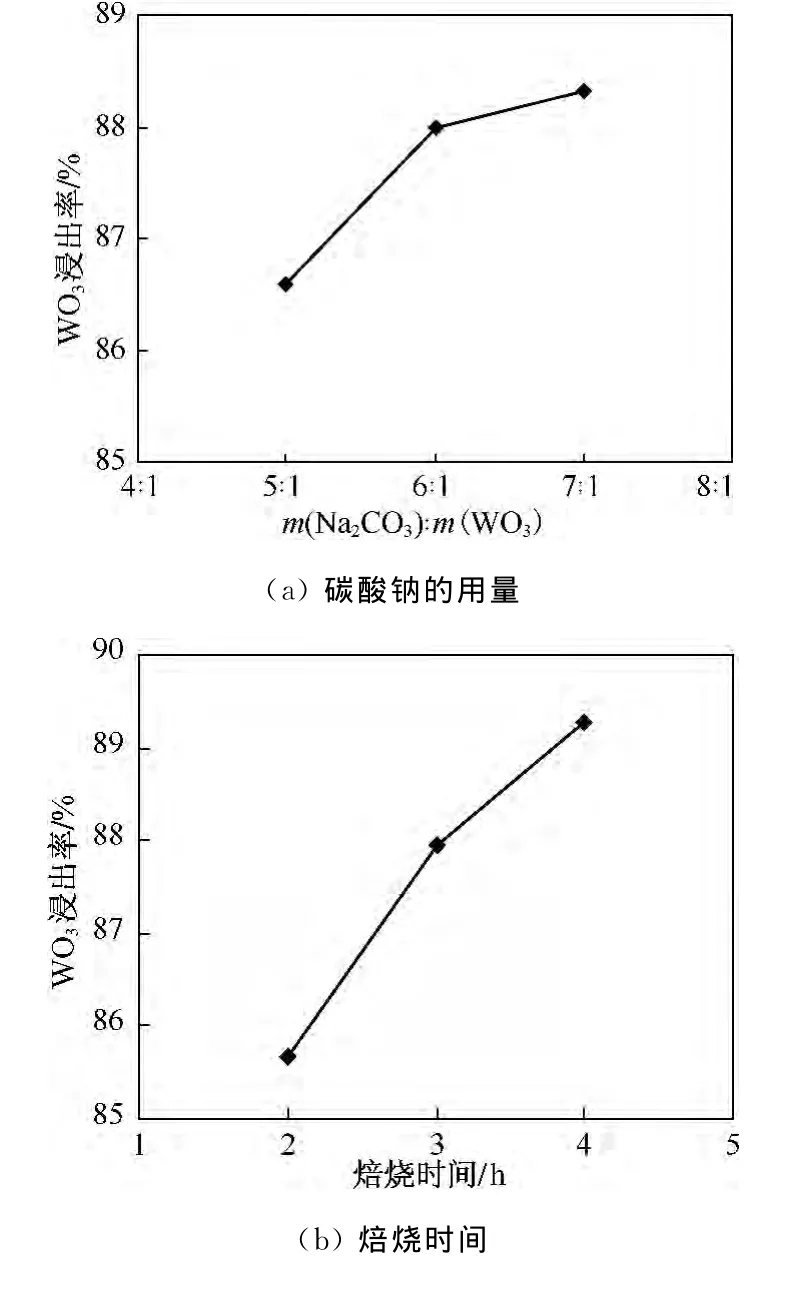
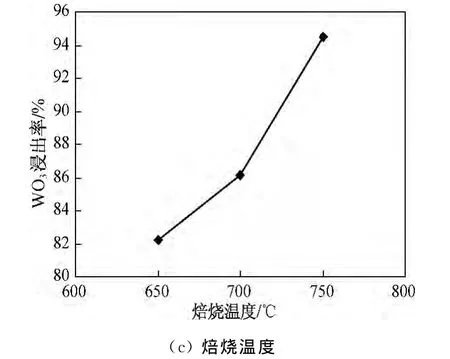
图1 焙烧试验各因素与WO3浸出率关系Fig.1 Effects of roasting factors on leaching rate of WO3
3.2 水浸试验条件的影响和分析
废催化剂在钠化焙烧正交试验得出的最优条件下焙烧,得到的焙料在500 r/min机械搅拌下恒温水浴中浸出,预备试验和条件试验确定了水浸出温度、水浸时间和固液比是影响WO3浸出率的主要因素以及各个因素的主要影响范围,对上述因素进行了正交试验,其结果如表3所示.

表3 水浸正交试验结果Table 3 Water leaching orthogonal experiments results
由表3的水浸正交试验结果得到水浸各因素对WO3浸出率的影响如图2所示.由图2可知,在水浸影响因素中,浸出温度对WO3浸出率的影响较为显著,当温度由90℃提高到95℃时,WO3浸出率上升不到1%,浸出时间和固液比的影响次之.因此,确定最佳的各因素水平组合:固液比为1∶4、浸出时间为2 h、浸出温度为90℃.
综合焙烧试验和水浸试验两部分,全程最佳各因素水平组合:焙烧温度为750℃、焙烧时间为4 h、m(Na2CO3)∶m(WO3)为6∶1、水浸温度为90℃、液固比为1∶4、浸出时间为2 h.在最佳因素水平下进行浸出试验得到WO3浸出率在95%以上.

图2 水浸试验各因素与WO3浸出率关系Fig.2 Effects of water leaching factors on leaching rate of WO3
3.3 影响镍浸出率的试验条件和因素分析
废催化剂经过焙烧水浸,过滤后的不溶物为富含氧化镍的原料,可采用酸浸对镍进行回收,在恒温水浴中进行酸浸,采用机械搅拌(可调速),将质量浓度为1.84 g/L的98%的浓硫酸稀释成不同质量分数的稀硫酸用于后续浸出试验.经预备试验和单因素条件试验确定了影响镍浸出率的主要因素为硫酸质量分数、浸出温度、固液比和浸出时间以及各个因素的主要影响范围.为了进一步得到镍浸出的最优化条件,找出各因素对镍浸出的显著影响性,对废催化剂经焙烧水浸后的过滤不溶物进行4因素3水平的L9(34)的酸浸正交试验,结果如表4所示.

表4 镍的酸浸正交试验结果Table 4 Acid leaching orthogonal experiments results of Ni
由表4的酸浸正交试验结果得到酸浸各因素对镍浸出率的影响如图3所示.由图3可知,酸浸各因素对镍浸出率的影响大小依次为:硫酸质量分数>浸出时间>浸出温度>固液比.但从固液比因素上看,当固液比由1∶8变为1∶9时,镍浸出率只提高了0.92%,增加液相已无法明显提高浸出率,表明1∶8的固液比足以达到了镍浸出的最佳比例;从硫酸质量分数影响来看,30%和40%的硫酸对镍浸出率差别很小,而硫酸的质量分数增大会造成物料的浪费,且增加了试验的难度和工艺实际操作的困难.因此,酸浸最佳各因素水平组合:浸出时间为4 h,浸出温度为85℃,硫酸质量分数为30%,固液比为1∶8.在该最佳条件下进行浸出试验,测得平均镍浸出率为98.27%.


图3 酸浸各因素对镍浸出率的影响Fig.3 Effects of acid leaching factors on leaching rate of Ni
4 浸出的动力学分析
4.1 浸出动力学模型
浸出过程的液-固相反应分3种类型:第一种是生成物可溶于水,此类反应可以用“未反应核收缩模型”描述;第二种是生成物为固态的浸出反应;第三种是固体反应物分散嵌布在惰性脉石基体中,液相反应物扩散到矿石内部致使浸出反应在矿石的表面和内部同时进行[10].本试验的硫酸浸镍过程属于第一种,即未反应核收缩模型.未反应核收缩模型控制步骤是由化学反应控制或扩散控制中最慢的一步决定,其动力学方程式[11-12]为
化学反应控制:1-(1-α)1/3=kt
其中:α为浸出率;t为反应时间;k为反应速率常数.
为了证实镍浸出过程的控制步骤符合哪种类型,本试验在硫酸质量分数为30%,固液比为1∶8,搅拌速度为500 r/min的条件下,研究了不同的温度下镍浸出率随时间的变化关系,如图4所示.由图4可以看出,反应温度对镍浸出率有较大影响,镍浸出率随着温度的增加而升高.
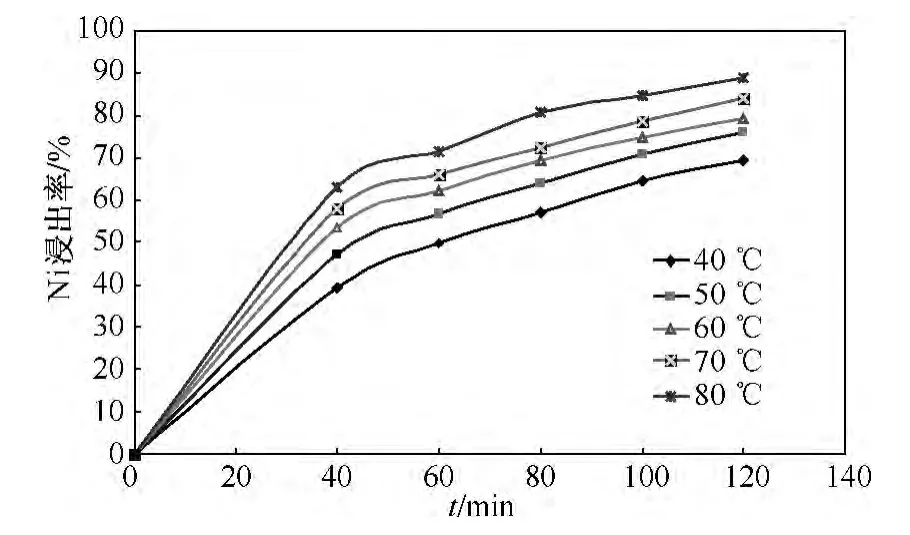
图4 不同的温度对镍浸出率的影响Fig.4 Effects of temperature factors on leaching rate of Ni
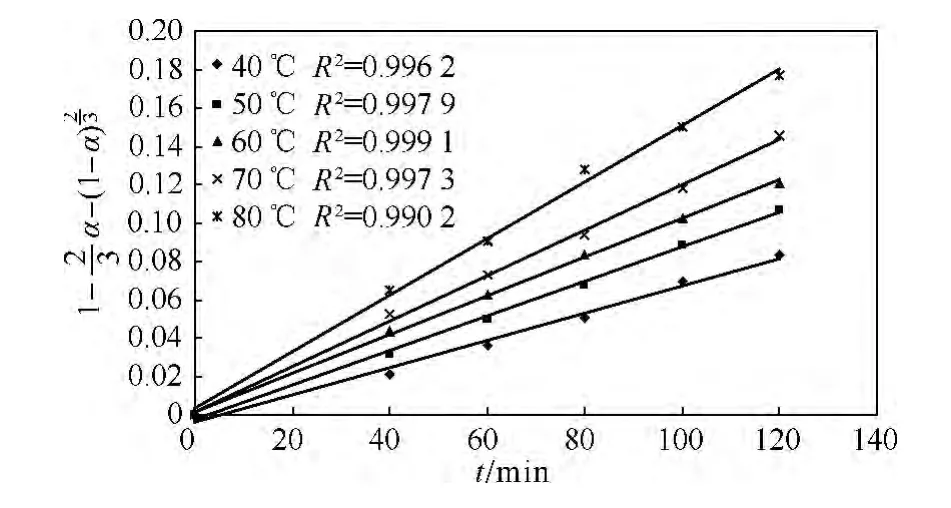
图5 不同温度下1-α- (1-α)2/3与浸出时间的关系Fig.5 1-α-(1-α)2/3 vs time at different temperatures
4.2 镍的酸浸反应活化能的计算
根据文献[11-15]报道,扩散控制的活化能一般小于25 kJ/mol,而化学控制的活化能一般大于40 kJ/mol.根据 Arrhenius方程k=A×e-E/RT(其中:k为反应速率常数,A为指前因子,E为表观活化能,R为摩尔气体常量,T为热力学温度),由图5可以求出各个直线的斜率即为不同温度下的反应速率常数k.以lnk对1/T作图,结果如图6所示,斜率为-E/RT,根据图6计算出镍的酸浸反应的活化能为15.95 kJ/mol.由此可知,硫酸浸出镍的表观活化能在典型的扩散控制活化能的范围之内,活化能的大小与前面推测的一致,进一步证明了镍的酸浸过程属于扩散控制模型.此反应活化能与文献[16]用硫酸浸出低品位硅锌矿计算的活化能(13.4 kJ/mol)及文献[17]用硫酸浸出废催化剂中的镍所计算得到的活化能(16.6 kJ/mol)相一致.
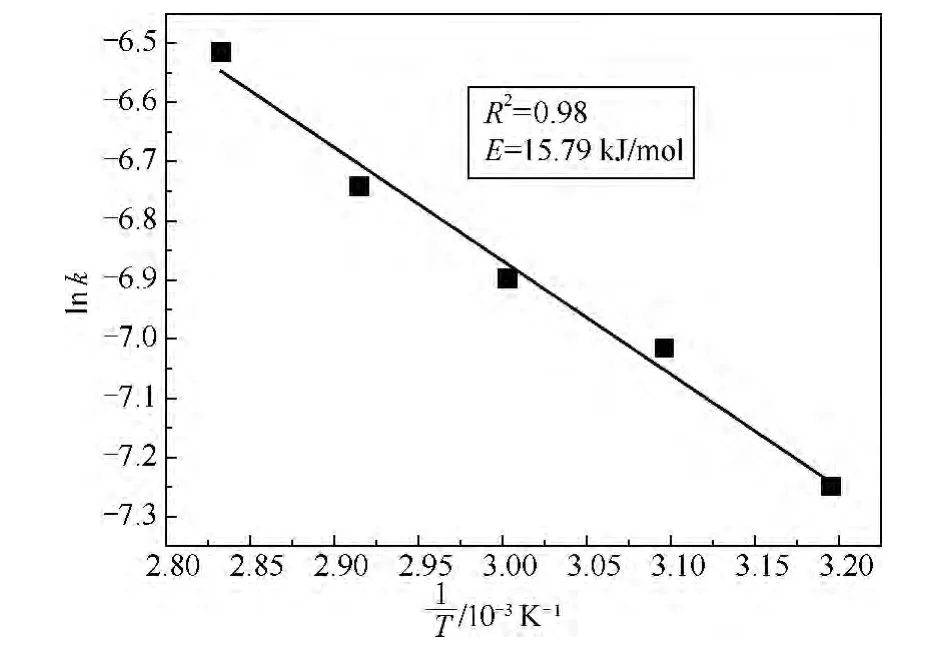
图6 ln k与1/T关系曲线Fig.6 Correlation of ln k vs 1/T
5 结 语
(1)通过对废催化剂的钠化焙烧和水浸的正交试验,得到了提取 WO3的最佳浸出条件:Na2CO3的用量是 WO3理论量的6倍、焙烧温度为750℃、焙烧时间为4 h、水浸温度为90℃、液固比为1∶4、浸出时间为2 h,在该最佳条件下 WO3的浸出率在95%以上.
(2)通过对镍的酸浸正交试验,得到了提取镍的最佳浸出条件:硫酸质量分数为30%、浸出时间为4 h、浸出温度为85℃、固液比为1∶8,在该最佳条件下镍的浸出率为98.27%.
(3)对镍的酸浸出动力学研究表明,该浸出过程属于扩散控制模型,浸出反应的表观活化能为15.95 kJ/mol.
[1]徐惠忠,王德义,赵鸣.固体废弃物资源化技术[M].北京:化学工业出版社,2004:126-131.
[2]陈祖庇.废催化剂的处理和利用[J].炼油技术与工程,2005,35(3):1-6.
[3]唐绍基.从废催化剂中回收有用金属[J].中国钨业,1996(3):15-17.
[4]崔燕,许民才.钴-钼废催化剂的回收利用现状及发展前景[J].安徽化工,2001,109(1):23-25.
[5]GHOLAMI R M,BORGHEI S M,MOUSAVI S M.Bacterial leaching of a spent Mo-Co-Ni refinery catalyst using acidithiobacillus ferrooxidans and acidithiobacillus thiooxidans[J].Hydrometallurgy,2011,106(1/2):26-31.
[6]LI Z,CHU Y C.A literature review of the recovery of molybdenum and vanadium from spent hydrodesulphurisation catalysts[J].Hydrometallurgy,2009,98(2):1-9.
[7]刘锦,蔡永红,任知忠,等.碱熔法回收废催化剂中的钴、钼、铝[J].化工环保,2004,24(2):134-137.
[8]黎庶,蔡萍,何伟.丁二酮肟光度法测定镍铁中镍[J].青海科技,2011(1):88-89.
[9]杭健,高若柳.硫氰酸盐比色法测定钨的改进[J].盐矿测试,2011,30(2):226-229.
[10]华一新.冶金过程动力学导论[M].北京:冶金工业出版社,2004:28.
[11]LEVENSPIEL O.Chemical reaction engineering[M].New York:Wiley,1972:367-383.
[12]HABASHI F.Principles of extractive metallurgy[M].New York:Gordon and Breach,1969:153-163.
[13]ABDEL-AAL E A,RASHAD M M.Kinetic study on the leaching of spent nickel oxide catalyst with sulfuric acid[J].Hydrometallurgy,2004,74(2):189-194.
[14]ANAND S,DAS S C,DAS R P,et al.Leaching of manganese nodules at elevated temperature and pressure in the presence of oxygen[J].Hydrometallurgy,1988,20(2):155-167.
[15]ROMANKIW L T, BRUYN D. Unit process in hydrometallurgy:Kinetics of dissolution of zinc sulfide in aqueous sulfuric acid[M]. Wadsworth:Dallas Tx,1963:62-85.
[16]ABDEL-AAL E A.Kinetics of sulfuric acid leaching of lowgrade zinc silicate ore[J].Hydrometallurgy,2000,55(3):247-254.
[17]MULAK W,MIAIQA B,SZYMLZYCHA A.Kinetics of nickel leaching from spent catalyst in sulfuric acid solution[J].International Journal of Mineral Processing,2005,77(4):231-235.
Recovery of Tungsten and Nickel from Spent Hydrogenation Catalyst and Acid Leaching Kinetics of Nickel
XUAo-yun,ZHENGCheng-zhi,ZHAOShu-hui
(College of Chemistry,Chemical Engineering and Biotechnology,Donghua University,Shanghai 201620,China)
The methods and the optimum process conditions of extraction of tungsten and nickel from spent hydrogenation catalysts were investigated by orthogonal experiments,while the leaching kinetics of nickel was also studied.The results showed that when the addition of Na2CO3was 5 times than theoretic WO3,the catalyst was sodium roasted under 750℃for 4 h,then leached in H2O under 90℃for 2 h,the leaching ratio of WO3could reach above 95%.The nickel was leached in sulfuric acid after being enriched in the leaching slag,a nickel recovery of 98%above was achieved with sulfuric acid of mass fraction of 30%,at 85℃for 4 h and the ratio of solid to liquid was kept at 1∶8.The leaching process of nickel was diffusion controlled model,which was consistent with diffusion controlled kinetic equation and the leaching activation energy was 15.95 kJ/mol.
spent catalyst;tungsten;nickel;diffusion controlled model;acid leaching kinetics
O 614
A
2013-05-17
许傲云(1989—),女,湖北天门人,硕士研究生,研究方向为废催化剂中有价金属的提取.E-mail:aoyunxu@163.com
赵曙辉(联系人),女,教授,E-mail:shzhao@dhu.edu.cn
1671-0444(2014)03-0300-06
