Mg-Er金属间化合物稳定性与电子结构的第一性原理研究
2014-06-04王文静刘子利刘希琴张志东王渠东
王文静,刘子利,刘希琴,张志东,王渠东
(1. 南京航空航天大学 材料科学与技术学院,南京 210016;2. 山东蒙沃变速器有限公司,临沂 273300;3. 上海交通大学 材料科学与工程学院,上海 200240)
作为最轻的工程金属材料,镁合金因其优异的力学性能和良好的铸造性、切削加工性、导热性、阻尼性以及较强的电磁屏蔽能力和易于回收等一系列优点,在航空航天、汽车和通讯工业上获得了广泛的应用。稀土元素RE是提高镁合金力学性能的重要合金化元素,RE与Mg具有较高的共晶温度(552~595 ℃),Nd、Ce、Ga和Er等稀土原子均能与Mg原子结合形成复杂晶体结构的金属间化合物,这些金属间化合物弥散分布在合金基体中,起到了很好的析出强化和细晶强化作用,能够显著提高镁合金的室温力学性能、耐热性能和铸造工艺性能[1−4]。
Mg-RE金属间化合物中RE与Mg原子的价键结合具有金属键和共价键两重性,直接影响了金属间化合物的力学性能和稳定性:当其中的金属键占主导地位时,其韧性比较好,稳定性较差;反之,则韧性较差,稳定性较好。利用电子理论相关的研究方法,从原子尺度上对化合物的成键方式进行研究分析,可以更好地了解Mg-RE二元化合物的力学性质、稳定性与其晶体结构、电子结构的内在联系。关于Mg-RE金属间化合物的电子理论研究工作已开展了很多,主要集中Nd、Ce、Ga、La等稀土上。WANG等[5]运用第一性原理方法系统研究了 Mg-Nd金属间化合物的结构稳定性,证明Mg-Nd合金中常温状态下的析出强化相应该是Mg41Nd5,而非Mg12Nd相。YANG等[6]、王殿辉等[7]研究了Mg-Ce体系中MgCe、Mg2Ce和Mg3Ce这3种金属间化合物的电子结构和弹性性质,得出了化合物的稳定性、弹性性质与晶体结构的内在联系机制。Mg-Ga合金体系中存在诸多金属间化合物相,其中 Mg5Ga2、Mg2Ga、MgGa、O-MgGa2(正交)、Mg2Ga5是稳定相,而H-MgGa2(六方)是亚稳相。GAO等[8]通过理论计算得出6种Mg-Ga金属间化合物的弹性性质和热力学性能,为相稳定性的研究提供了新方法。Mg-La合金体系中存在 5种金属间化合物:MgLa、Mg2La、Mg3La、Mg17La2和 Mg12La,随着 Mg-La化合物中 La含量的下降,费米能级低能级区域成键电子数减少,化合物形成能的绝对值减小,稳定性降低[9]。
Er属于重稀土金属,在 Mg中的最大固溶度(32.7%)远高于在其他常见稀土元素的,因此,Er对镁合金的性能影响效果比常用的轻稀土元素更为显著,含 Er镁合金的研究受到了极大的关注[10−13]。关于含Er镁合金的研究主要集中在力学性能和耐腐蚀性能方面,目前尚未见关于Mg-Er金属间化合物的电子理论方面的研究报道。本文作者采用基于密度泛函理论的第一性原理计算方法优化了 MgEr、Mg2Er和Mg24Er5这3种Mg-Er金属间化合物的晶体结构,通过形成热、结合能的计算并结合电子结构分析了金属间化合物的稳定性与微观结构、原子键合方式之间的关系。
1 模型与计算方法
1.1 晶体结构与模型
根据Mg-Er二元相图,室温下Mg-Er合金中存在MgEr、Mg2Er和Mg24Er5这3种金属间化合物,这3种金属间化合物结构参数如表 1所列[14−17]。MgEr中各原子的坐标分别如下:Er(0,0,0),Mg(0.5,0.5,0.5)。Mg2Er中各原子的坐标分别如下:Er(0.333 3,0.666 7,0.063 0);Mg(0,0,0)、(0.833 0,0.666 0,0.250 0)。Mg24Er5中各原子的坐标分别如下:Er(0,0,0)、(0.312 6,0.312 6,0.312 6);Mg(0.355 7,0.355 7,0.032 4)、(0.092 7,0.092 7,0.279 1)。3 种化合物的结构模型分别如图1所示。
1.2 计算方法
采用基于密度泛函理论的程序软件包 CASTEP(Cambridge serial total energy package)来进行3种模型的相关计算[18]:选择广义梯度近似 GGA(Generalized gradient approximation)下的 PBE(Perdew-Burke-Ernzerhof)泛函来处理交换关联能[19],价电子所处的势场用超软赝势来构造,自洽场运算时采用Pulay密度混合法,倒易空间中平面波计算的最大截止能量Ecut为380.0 eV,MgEr、Mg2Er晶胞Brillouin区的K点取为5×5×2,Mg24Er5的取为2×2×2,自洽精度设为1×10−6eV/atom。结构优化结束时,体系总能量收敛到2×10−5eV/atom,每个原子上的力低于0.5 eV/nm,公差偏移为2×10−4nm,应力偏差为0.1 GPa。考虑到计算所涉及的原子包含稀土原子 Er,它具有特殊 4f核外电子层,对模型进行与能量相关的计算时要对Er的f电子层进行HubbardU设置[20],U设为6。
在金属间化合物的形成热和结合能计算中将涉及到 Mg、Er单个原子固态以及孤立态时的能量。表 2列出了Mg、Er原子的晶体类型、固态单原子能量计算时K点和截断能Ecut的设置、孤立态单原子能量计算时截断能Ecut的设置、单个原子固态以及孤立态的能量。计算孤立态单原子能量时,构建晶格常数为10 Å的简单立方结构模型,将单个原子置于模型的中心位置,K点设为Gamma点。

表1 Mg-Er金属间化合物结构参数[14−17]Table 1 Structure parameters of Mg-Er intermetallic compounds[14−17]
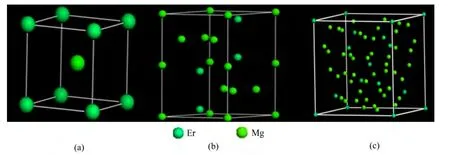
图1 Mg-Er金属间化合物晶胞模型Fig. 1 Cell models of Mg-Er intermetallic compounds: (a) MgEr; (b) Mg2Er; (c) Mg24Er5
2 计算结果与讨论
2.1 晶格常数
表3所列为对MgEr、Mg2Er和Mg24Er5晶胞体积及原子位置优化计算后的平衡晶格常数和晶胞总能Etot。MgEr、Mg2Er和 Mg24Er5的晶格常数与文献[14−17]给出的实验值小5%,表明优化计算结果可靠。
2.2 形成热与结合能
形成热是指原子由单质状态形成化合物时释放的能量,可用于表征金属间化合物形成的难易程度,当形成热为负值时,其绝对值越大,表示此金属间化合物越易形成[21−23]。Mg-Er金属间化合物的形成热H为[24]
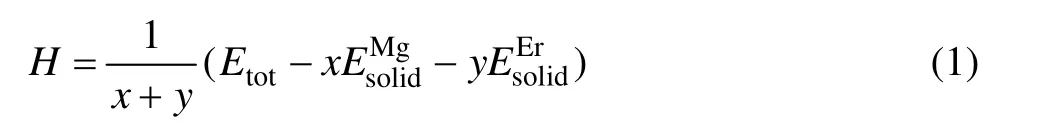
式中:x、y分别表示金属间化合物晶胞结构中 Mg、Er原子的原子个数;Etot表示化合物晶胞结构优化完成后的总能量;分别表示单个 Mg、Er原子在固态时的能量。在计算固态单原子能量、时,采用与金属间化合物晶胞总能量相同的计算条件,分别为对Mg、Er原子相应的单质能量进行计算,然后除以单质中总的原子个数后所得的能量,即Mg、Er的固态单原子能量。Mg原子与Er原子的固态单原子能量分别为−973.951 eV/atom 和−5 224.094 eV/atom(表2),结合表3中3种化合物的总能数据,代入式(1)计算得出MgEr、Mg2Er和Mg24Er5的形成热分别为−1.386、−1.084和−0.214 eV/atom。可以看出,3种化合物的形成热均为负值,其中MgEr形成热的绝对值最大,形成能力最强,Mg2Er次之,Mg24Er5的形成能力最差(见图2(a))。

表2 固态及孤立态单个原子的能量Table 2 Energies of solid and isolated atom
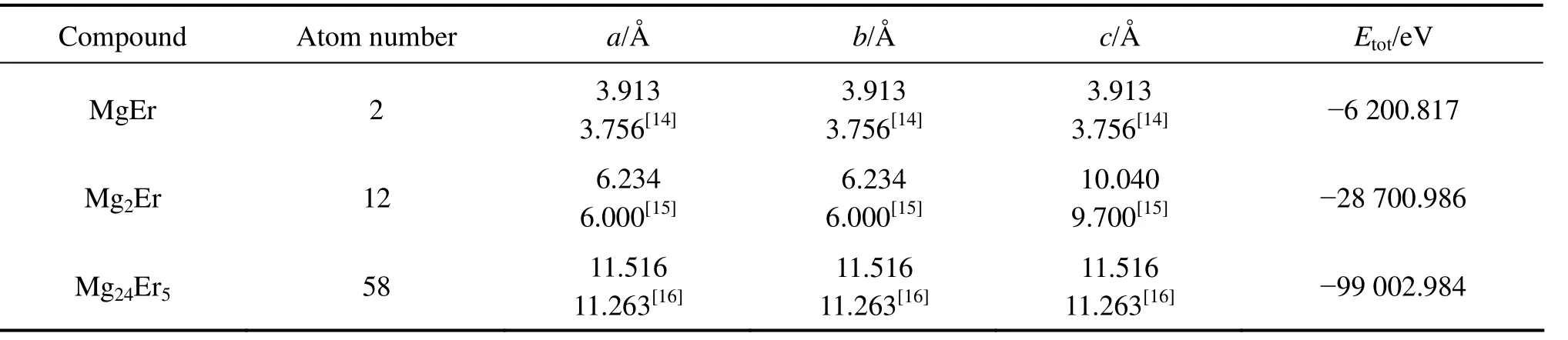
表3 MgEr、Mg2Er和Mg24Er5的平衡晶格常数与晶胞总能Table 3 Equilibrium lattice constants and total energy of MgEr, Mg2Er and Mg24Er5
晶体结合能是将自由原子结合为晶体所释放的能量,也就是把晶体分解成单个原子时外界所做的功。晶体的强度、结构稳定性与其结合能密切相关,当结合能为负值时,其绝对值越大,则形成的晶体就越稳定[21,23,25]。Mg-Er金属间化合物结合能Ecoh为[24]
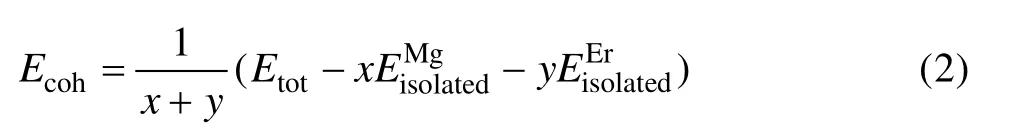
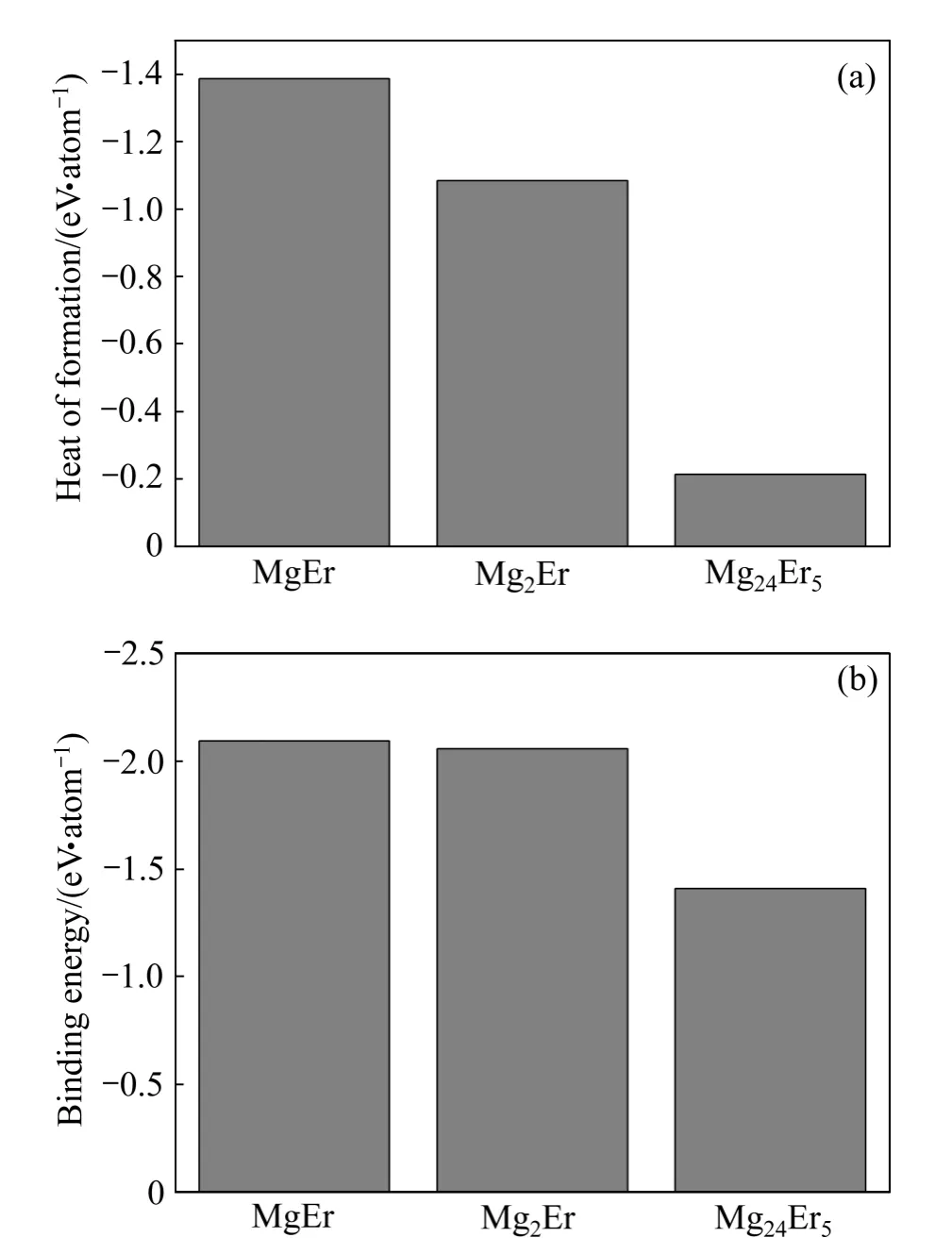
图2 MgEr、Mg2Er和Mg24Er5的形成热和结合能Fig. 2 Heat of formation(a) and binding energy(b) of MgEr,Mg2Er and Mg24Er5
2.3 态密度
图3 所示为3种Mg-Er金属间化合物的分波态密度(PDOS)和总态密度(TDOS)图。由 3种化合物的总态密度图可以看出,费米能级处的电子数不为零,说明这3种化合物均呈明显的金属特性。3种化合物的总态密度图中成键电子的分布区间基本一致,主要集中在4个能量区间:−47~−49 eV,−43~−44 eV,−21~−23 eV和−6~0 eV。−47~−49 eV价带区的电子主要由Er的6s轨道贡献;−43~−44 eV价带区的电子由Mg的2p轨道贡献;−21~−23 eV价带区的电子由Er的5p轨道贡献;在−6~0 eV价带区,Mg的3s、2p轨道与Er的4f、5d轨道发生重叠,说明在该区域产生了轨道杂化,Mg-Er金属间化合物的主要成键峰分布在−6~0 eV的范围内。Mg的2p轨道与Er的4d轨道在导带区也有一定量的杂化。费米能级低能级区域的电子是主要的成键电子,在费米能级附近区域原来空的 Er 5d态由于电荷转移变得被部分占据(部分来自 Er 6s,部分来自Er 4f)[27],因而,Er 5d电子对金属间化合物中原子间的成键也有贡献。
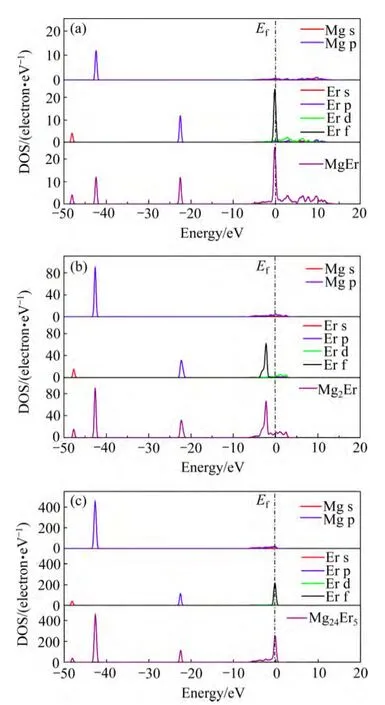
图3 MgEr、Mg2Er和Mg24Er5的分波态密度(PDOS)和总态密度(TDOS)图Fig. 3 Partial and total DOS of MgEr(a), Mg2Er(b) and Mg24Er5(c)
底带宽度的大小能够反映出电子局域程度的强弱和电子参与成键能力的大小:底带宽度小,电子局域程度强,原子轨道扩展性弱,电子参加成键的能力小;反之,则电子局域程度弱,原子轨道扩展性强,电子参加成键的能力大。在PDOS图中可以看到,Mg的s、p轨道决定了费米面附近的底带宽度。Mg-Er 3种化合物的底带能级分别在−6~12,−6~3,−6~1 eV之间。对比3种化合物的TDOS图,可以看出,随着化合物中Er含量的增加,3种化合物在费米能级附近的底带宽度逐渐增大,且主要表现在费米能级右边高能区域宽度的增大。因此,Er元素的加入提高了电子参加成键的能力,化合物的非金属性和稳定性随Er含量的升高而增强。
在合金体系中,合金元素的平均成键电子数能很好地反映合金的稳定性。成键电子主要位于费米能级以下,当有更多的电子处在较低能量时,成键电子数增加,晶体内的价电子的相互作用增强,晶体的稳定性将增大[28]。使用电子态密度积分的方法计算出平均每个原子在费米能级低能级处的成键电子数,以此为判据,可以判断出合金中各个化合物稳定性的高低[29−30]。Mg-Er合金体系中,MgEr、Mg2Er和 Mg24Er5中平均每个原子在费米能级低能级处的成键电子数分别为7.38、5.99和3.82。可以看出,随着Er含量的减少,各个金属间化合物平均每个原子在费米能级低能级处的成键电子数减少,3种化合物的稳定性随Er含量的降低是逐渐减小的,这与上述结合能计算得出的结论一致。
2.4 电荷密度与差分电荷密度
图4所示为3种Mg-Er金属间化合物(110)面的电荷密度图。由电荷密度图可以看出,每个原子周围的电荷均呈球形分布,在Mg、Er原子周围均有大量的电荷存在,呈典型的金属键特征。
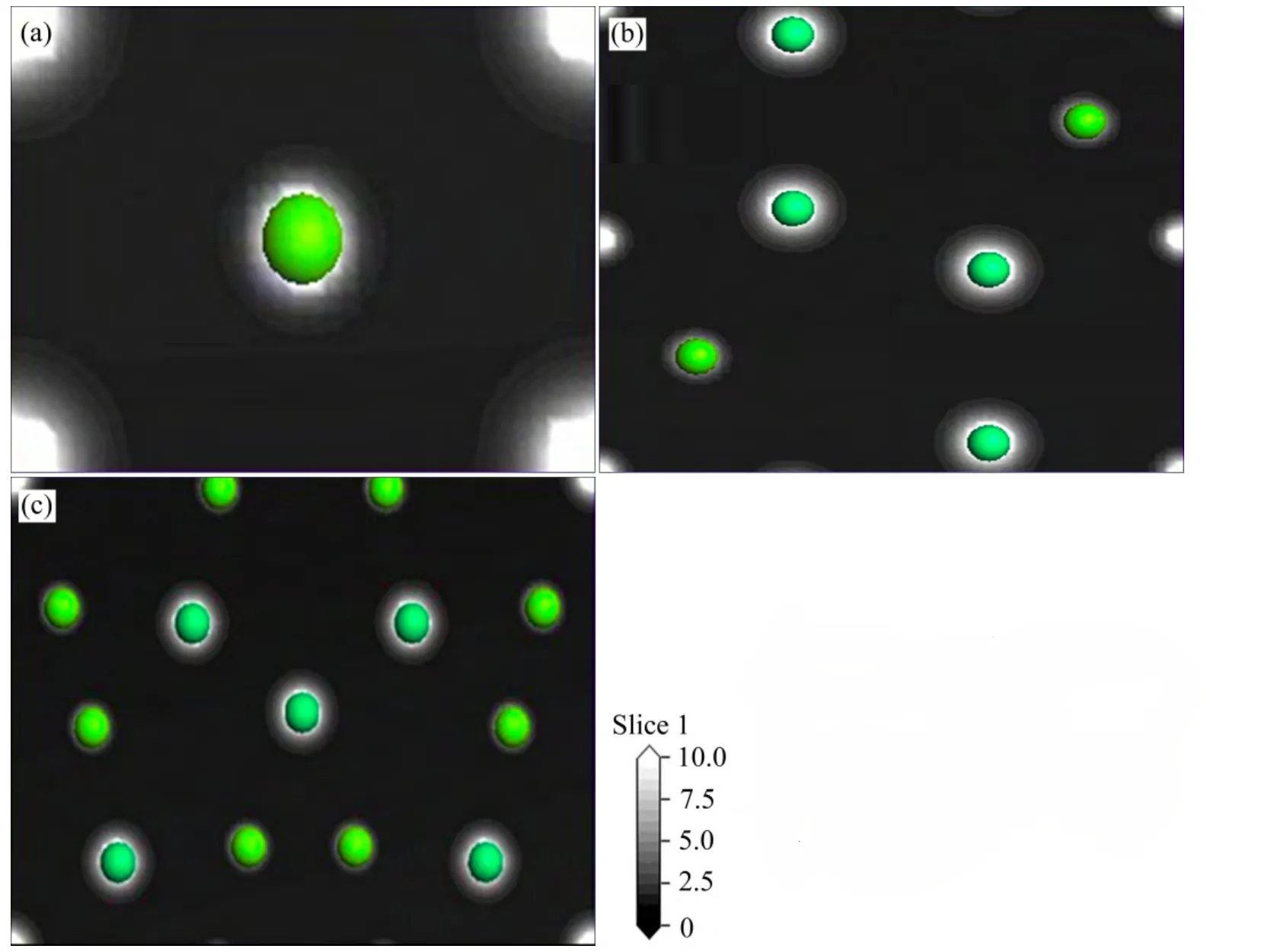
图4 MgEr、Mg2Er和 Mg24Er5的(110)面电荷密度图Fig. 4 Charge densities on (110)plane of MgEr(a), Mg2Er(b) and Mg24Er5(c)

图5 MgEr、Mg2Er和Mg24Er5的(110)面差分电荷密度图Fig. 5 Charge densities difference on (110) plane of MgEr(a), Mg2Er(b) and Mg24Er5(c)
图5所示为 3种化合物(110)面的差分电荷密度图,电子密度采用红、白、蓝标示,蓝色表示得电子区域,红色为失电子区域,白色是中间态。可以看出,3种化合物的(110)面上的Er和Mg原子周围出现了电荷转移,Er原子和Mg原子位置为失电子区域,电荷密度差为负值,而它们之间为得电子区域,电荷密度差为正值,表明 Mg原子与 Er原子存在共用电子,Mg-Er原子间形成了方向性较强的共价键。由图3中态密度分析可知,Mg、Er原子间存在少量的轨道杂化,而由差分电荷密度图可以看出,Mg、Er之间的电子云只有部分重叠,交界处电荷的畸变不大。所以,Mg-Er金属间化合物的价键结合具有金属键和共价键两重性,其中金属键占主导地位,共价键所占比例较小。对比MgEr和Mg2Er两种化合物的差分电荷密度图可以看出(见图5),MgEr中Mg原子与Er原子之间的电荷转移量高于 Mg2Er中两原子之间的电荷转移量,这表明 MgEr中 Mg-Er方向共价键的强度高于Mg2Er中的。共价键强度越高,则化合物的稳定性越好[31],所以,MgEr的稳定性要比Mg2Er的高,这与上述结合能和态密度所得结论一致。在 Mg24Er5的差分电荷密度图中Er原子密集分布的地方,原子间电荷转移量较大,说明在这些区域共价键性比较强;而在Er原子分布较少的区域,原子间电荷转移量较少,说明在这些区域共价键性比较弱。由于 Mg24Er5晶胞中Er原子含量较低,所以,在相同原子数的3种Mg-Er化合物的晶胞中,Mg24Er5中 Mg-Er共价键的数量最少,总体的电荷转移量最小,稳定性最差。
3 结论
1) Mg-Er金属间化合物 MgEr、Mg2Er、Mg24Er5的形成热和结合能均为负值,并且随着化合物中 Er含量的降低,化合物的形成能力和稳定性都降低。
2) 随着Er含量的降低,金属间化合物平均每个原子在费米能级低能级区域的成键电子数随之减少,晶体中价电子的相互作用减弱,化合物的稳定性降低。
3) 在Mg、Er原子周围有大量的电荷存在,呈典型的金属键特征。Mg、Er之间存在共用电子,形成了方向共价键,但交界处电荷的畸变不大。Mg-Er金属间化合物的价键结合具有金属键和共价键两重性,其中金属键占主导地位。Mg、Er原子的电荷转移量随化合物中Er含量降低而减少,化合物的共价键性降低,稳定性下降。
[1]WANG Qu-dong, LU Yi-zhen, ZENG Xiao-qin, DING Wen-jiang, ZHU Yan-ping. Effects of RE on microstructure and properties of AZ91 magnesium alloy[J]. Transactions of Nonferrous Metals Society of China, 2000, 10(2): 235−239.
[2]XIN Ren-long, LI Li, ZENG Ke, SONG Bo, LIU Qing.Structural examination of aging precipitation in a Mg-Y-Nd alloy at different temperatures[J]. Materials Characterization, 2011,62(5): 535−539.
[3]张诗昌, 魏伯康, 林汉同, 王立士. 钇及铈镧混合稀土对AZ91镁合金铸态组织的影响[J]. 中国有色金属学报, 2001,11(S2): 99−102.ZHANG Shi-chang, WEI Bo-kang, LIN Han-tong, WANG Li-shi. Effect of yttrium and mischmetal on as-cast structure of AZ91 alloy[J]. The Chinese Journal of Nonferrous Metals, 2001,11(S2): 99−102.
[4]刘楚明, 葛位维, 李慧中, 陈志永, 王 荣, 高艳蕊. Er对铸态 AZ91镁合金显微组织和耐腐蚀性能的影响[J]. 中国有色金属学报, 2009, 19(5): 847−853.LIU Chu-ming, GE Wei-wei, LI Hui-zhong CHEN Zhi-yong,WANG Rong, GAO Yan-rui. Effect of Er on microstructure and corrosion resistance of AZ91 magnesium alloy[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(5): 847−853.
[5]WANG Can, HAN Pei-dei, ZHANG Lu, ZHANG Cai-li, XU Bing-she. First-principles study on the stabilities of the intermetallic compounds in Mg-Nd alloys[J]. Rare Metal Materials and Engineering, 2011, 40(4): 590−594.
[6]YANG Fang, WANG Ji-wei, KE Jiang-ling, PAN Zheng-gui,TANG Bi-yu. Elastic properties and electronic structures of Mg-Ce intermetallic compounds from first-principles calculations[J]. Physica Status Solidi B, 2011, 248(9):2097−2102.
[7]王殿辉, 王仲民, 吉宗威, 胡朝浩, 周怀营. Ce-Mg合金体系弹性性质与电子结构的第一性原理研究[J]. 桂林电子科技大学学报, 2012, 32(4): 339−344.WANG Dian-wu, WANG Zhong-min, JI Zong-wei, HU Chao-hao, ZHOU Huai-ying. First-principle study of elastic properties and electronic structure of Ce-Mg alloys[J]. Journal of Guilin University of Electronic Technology, 2012, 32(4):339−344.
[8]GAO Qian-nan, DU Yong, ZHAO Dong-dong, WANG Ai-jun,WANG Jiong, LIU Shu-hong, QU Yi-fang. Elastic, phonon and thermodynamic properties of Mg-Ga compounds from first-principles calculations[J]. Computer Coupling of Phase Diagrams and Thermochemistry, 2012, 37: 137−144.
[9]WANG Yu-fei, ZHANG Wei-bing, WANG Zhi-zhong, DENG Yong-he, YU Na, TANG Bi-yu, ZENG Xiao-qin, DING Wen-jing. First-principles study of structural stabilities and electronic characteristics of Mg-La intermetallic compounds[J].Computational Materials Science, 2007, 41: 78−85.
[10]邱从章, 刘楚明, 常亚喆, 李慧中, 王孟君. 铸态Mg-Zn-Er-Zr合金显微组织和力学性能的研究[J]. 材料热处理技术, 2008, 37 (14) : 39−41.QIU Cong-zhang, LIU Chu-ming, CHANG Ya-zhe, LI Hui-zhong, WANG Meng-jun. Research on microstructure and mechanical properties of Mg-Zn-Er-Zr alloy as-cast[J]. Material and Heat Treatment, 2008, 37(14): 39−41.
[11]张晋涛, 陈乐平, 尹 健, 周 全. 微量Er对ZM5镁合金微观组织及腐蚀性能的影[J]. 铸造技术, 2012, 33(2): 160−163.ZHANG Jin-tao, CHEN Ping-le, YIN Jian, ZHOU Quan.Influence of Er addition on microstructure and corrosion resistance of ZM5 magnesium alloy[J]. Foundry Technology,2012, 33(2): 160−163.
[12]WANG Zhong-jun, XU Yang, ZHU Jing. Effects of erbium addition on the corrosion resistance and microstructure of AZ91 magnesium alloy[J]. Advanced Materials Research, 2011,194/196: 1221−1224.
[13]张 静, 袁付庆, 豆雨辰, 张旭峰, 汤爱涛. 稀土 Er对 ZK21镁合金组织的影响[J]. 材料热处理学报, 2011, 32(11): 94−98.ZHANG Jing, YUAN Fu-qing, DOU Yu-chen, ZHANG Xu-feng,TANG Ai-tao. Effects of rare earth Er on microstructure of ZK21 alloy[J]. Transactions of Materials and Heat Treatment, 2011,32(11): 94−98.
[14]BUSCHOW K H J. Magnetic properties of CsCl-type rare earth-magnesium compounds[J]. Journal of the Less Common Metal, 1973, 33(2): 239−244.
[15]KRYPYAKEVICH P I, EVDOKIMENKO V I, ZALUTSKII I I.Hexagonal Laves phases in the alloys of magnesium with rare-earth metals[J]. Reports of the Academy of Sciences of the Ukrainian SSR, 1964, 6: 766−769
[16]SCHÄFER W, BUSCHOW K H J. Neutron diffraction study on intermetallic Er5Mg24and Tm5Mg24[J]. Materials Science Forum,2004, 443: 263−266.
[17]SACCONE A, DELFINO S, MACCIO D, FERRO R. Phase equilibria in the binary rare-earth alloys: The erbium-magnesium system[J]. Metallurgical Transactions A, 1992, 23(3):1005−1012.
[18]SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J,HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. Journal of-Physics: Condens Matter, 2002, 14: 2717−2743.
[19]PERDEW J P, CHEVARY J A, VOSKO S H, JACKSON K A,PEDERSON M R, SINGH D J. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation[J]. Physical Chemistry B, 1992, 46:6671−6687.
[20]ANISIMOV V I, ZAANEN J, ANDERSEN O K. Band theory and Mott insulators: Hubbard U instead of Stoner I[J]. Physical Review B, 1991, 44: 943−954.
[21]WU Meng-meng, WEN Li, TANG Bi-yu, PENG Li-ming, DING Wen-jiang. First-principles study of elastic and electronic properties of MgZn2and ScZn2phases in Mg-Sc-Zn alloy[J].Journal of Alloys and Compounds, 2010, 506: 412−417.
[22]FU C L, WANG X D, YE Y Y, HO K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3by first-principle[J].Intermetallics, 1999, 7(2): 179−184.
[23]CHEN Lü, PENG Ping, LI Gui-fa, LIU Jin-shui, HAN Shao-chang. First-principle calculation of point defective structures ofB2-RuAl intermetallic compound[J]. Rare Metal Materials and Engineering, 2006, 35(7): 1065−1070.
[24]SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Materials Science and Engineering B, 1997, 49(1):74−78.
[25]SONG Y, GUO Z X, YANG R, LI D. First principles study of site substitution of ternary elements in NiAl[J]. Acta Materialia,2001, 49(9): 1647−1654.
[26]吴玉蓉. 稀土镁合金的热力学及固溶特性的理论模拟[D]. 长沙: 湖南大学, 2007: 72−81.WU Yu-rong. Theory simulation for thermodynamic and solid solution properties of eare magnesium alloys[D]. Changsha:Hunan University, 2007: 72−81.
[27]马兰春. 含局域d/f电子的化合物电子结构的第一性原理研究[D]. 上海: 复旦大学, 2007: 75.MA Lan-chun. First-principles studies on electronic structures of compounds containing local d/f electrons[D]. Shanghai: Fudan University, 2007: 75.
[28]GHOSH G. First-principles calculations of structural energetics of Cu-TM (TM=Ti, Zr, Hf) intermetallics[J]. Acta Materialia,2007, 55: 3347−3374.
[29]YU R, HE L L, YE H Q. Effect of W on structural stability of TiAl intermetallics and the site preference of W[J]. Physical Review B, 2002, 65(18): 184102−184107.
[30]NYLÉN J, GARCÌA F J G, MOSEL B D, PÖTTGEN R,HÄUSSERMANN U. Structure relationships, phase stability and bonding of compounds PdSnn(n=2, 3, 4)[J]. Solid State Sciences,2004, 6(1): 147−155.
[31]李燕峰, 徐 慧, 张 彪, 章立刚. Al-Sc金属间化合物的电子结构及稳定性和热力学性质[J]. 中国有色金属学报, 2010,20(5): 946−953.LI Yan-feng, XU Hui, ZHANG Biao ZHANG Li-gang.Electronic structure, stability and thermodynamic properties of Al-Sc intermetallics compounds[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(5): 946−953.
