新城疫病毒NP基因的克隆与原核表达研究
2014-05-30刘苏君高永安张陈量金俊杰涂宜强荀飞琼
刘苏君,高永安,张陈量,金俊杰,涂宜强,荀飞琼,2,白 宇*
(1.温州科技职业学院,浙江温州325006;2.温州欧斯达康生物公司)
新城疫(ND)是由新城疫病毒(NDV)引起的一种禽类急性、高度接触性传染病,给养禽业带来严重的经济损失[1-2]。
NDV 为一种有囊膜、单股不分节段的负链RNA病毒。NDV 基因组全长约15 186 bp,编码6 个主要病毒结构蛋白:核衣壳蛋白(NP)、磷蛋白(P)、基质蛋白(M)、融合蛋白(F)、血凝素-神经氨酸酶蛋白(HN)和大分子蛋白(L)[3]。其中,NP 蛋白是组成病毒核衣壳的主要结构蛋白,与L 蛋白、P 蛋白包裹病毒基因组RNA 形成螺旋状的核糖壳,在转录和复制过程中充当模板,NP 蛋白N 末端2/3 与RNA 直接结合,并作为核衣壳组装所必须的区域[4]。NP蛋白还具有较高的免疫原性,可能与其刺激机体产生免疫机能有关[5-6]。
本试验旨在克隆NDV LaSota 毒株NP 基因,利用原核表达系统获得纯化的NP 蛋白,为进一步研究其生物学特性和制备相应抗体奠定基础。
1 材料与方法
1.1 病毒、载体与菌株 NDV 毒株为鸡新城疫活疫苗(LaSota 株),购自哈尔滨维科生物技术开发公司。
pBluscript ⅡKS(+ /-)和pET-28a 载体由温州科技职业学院动物科学系动物病毒研究室保存,感受态细胞E coli DH5α 和BL21(DE3)工程菌购自某生物工程(大连)有限公司。
1.2 工具酶及主要试剂 RNA 酶抑制剂、PCR 反应试剂、DNA Marker(DL2000、DLl5000)、PrimeSTAR HS DNA 聚合酶、T4 DNA 连接酶、鼠源反转录酶(MMLV)、IPTG、限制性内切酶(Eco R I,Xho I,Eco R V)购自某生物工程(大连)有限公司;DNA 胶回收试剂盒、质粒小提试剂盒购自某生物工程(上海)股份有限公司;Ni-NTA 纯化树脂购自QIAGEN 公司。
1.3 引物设计与合成 本试验所用引物以Gene Bank 上公布的NDV LaSota 株基因组序列(基因序列号为AF077761)为参考序列,利用Primer 5.0 软件进行设计,由某生物工程(上海)股份有限公司合成。
上游引物P1:
5'-CCGGAATTCATGTCTTCCGTATTTGA-3'
下游引物P2:
5'-CCGCTCGAGTCAATACCCCCAGTC-3'
为便于构建表达载体,在上、下游引物5'端分别加入Eco R I 和Xho I 酶切位点(引物中下划线部分),并加3 个保护性碱基。
1.4 NDV RNA 提取与反转录 NDV LaSota 株疫苗病毒液150 μL,参照说明书用试剂盒提取病毒RNA。然后进行反转录,合成病毒cDNA。
反转录体系如下:先将RNA 22 μL:特异性下游引物1 μL;dNTP(2.5 mm)3 μL;DTT 3 μL;65℃5 min;冰水浴5 min;然后加入5 × FS Buffer 8 μL;RNase Inhibitor 1 μL;M-MLV 1 μL 37℃水浴2 h,-20℃保存备用。
1.5 目的基因的PCR 扩增与克隆 以反转录得到NDV LaSota 株NP 基因cDNA 为模板,利用特异引物进行PCR 扩增。
PCR 体系:5 ×primestar Buffer 10 μL;primestar 0.5 μL;dNTP(2.5 mm)4 μL;P1(上游引物)1.5 μL;P2(下游引物)1.5 μL;Template(cDNA)4 μL;dH2O 28.5 μL。
反应条件:95℃预变性5 min,98℃变性10 sec,56℃退火15 sec,72℃延伸1.5 min,反应30 个循环后,72℃终延伸10 min,反应结束后,对PCR 产物进行核酸电泳鉴定。
NDV LaSota 株NP 基因克隆:将上述PCR 产物胶回收后与pBluscript ⅡKS(+ /-)载体16℃连接2 h。构建重组质粒,命名为pBlue-NDV-NP。将pBlue-NDV-NP 转入感受态DH-5α 中并均匀涂在氨苄抗性的LB 平板上。经IPTG 诱导,17 h 后挑取菌落中白色阳性菌落,37℃培养16 h。参照试剂盒说明书提取质粒,将初步鉴定阳性质粒送某生物工程(上海)股份有限公司进行测序。
1.6 NDV NP 基因表达栽体的构建与鉴定 经酶切和测序鉴定的载体pBlue-NDV-NP 用Eco R I 和Xho I 进行双酶切处理。胶回收试剂盒回收获得NP基因。
pET-28a 载体用Eco R I 和Xho I 进行双酶切处理,与回收获得的NP 基因进行连接后,转入DH-5α并均匀涂在含卡那霉素抗性的LB 平板上。重组质粒pET-28a-NDV-NP 用Eco R I 、Xho I 酶切鉴定。鉴定正确后转入感受态BL21(DE3)用于表达。
1.7 重组质粒的诱导表达和重组蛋白的可溶性分析 设置空载体pET-28a 为对照。用含卡那霉素的新鲜LB 液体培养基按1∶100 比例分别稀释pET-28a-NDV-NP 与空载体pET-28a 菌液,37℃震荡培养至菌液OD600值达0.6 时,用终浓度为1.0 mM/L IPTG 37℃诱导5h。取15 mL 后菌液,6000 g 离心10 min,去上清,加入结合缓冲液重悬沉淀。
经超声处理后,6000 g 离心分离上清与沉淀样品,用于SDS-PAGE 检测,确定重组蛋白是否为可溶性表达。
1.8 表达产物的SDS-PAGE 分析 将收集到的菌液4℃,12000 r/min 离心10 min 后收集沉淀,PBS重悬,加入等体积2 ×SDS 凝胶上样缓冲液,煮沸5 min,作为SDS-PAGE 电泳上样样品。
按照分子克隆实验指南方法进行SDS-PAGE 电泳[7],浓缩胶5%,分离胶12%,恒压120 V,同时以含空载体菌体和含重组阳性载体诱导前菌体为对照。电泳完毕后,取下凝胶,置于考马斯亮兰R250染色,脱色液脱色数次后分析蛋白的表达情况。
1.9 重组蛋白的纯化与Western-blot 分析 取100 mL 诱导后的菌液,10000 g 4℃离心5 min,弃上清。将细菌沉淀中加入结合缓冲液重悬后。超声破碎细胞,10000 g 离心20 min,收集上清。参照QIAGEN纯化试剂盒说明进行蛋白纯化,纯化样品进行SDSPAGE 检测,检测纯化效果。
将SDS-PAGE 凝胶电泳蛋白条带转印到NC 膜上,用洗涤液PBST(含有0.05%吐温20)洗膜3 次,每次3 min;然后加入抗His-tag 单抗为一抗(1∶2000稀释),室温振荡1 h;再用PBST 洗膜3 次,每次3 min;然后加入1∶5000 稀释的辣根过氧化酶(HRP)标记的羊抗鼠-抗体为二抗,室温振荡1.5~2 h;再用PBST 漂洗3 次,每次3 min;最后置于DAB 显色液中显色,待条带清晰后迅速用dH2O 终止显色。
2 结果与分析
2.1 NDV LaSota 株NP 基因的克隆和pBlue-NDVNP 构建与酶切鉴定 详见图1、图2。

图1 NDV NP 基因PCR 扩增
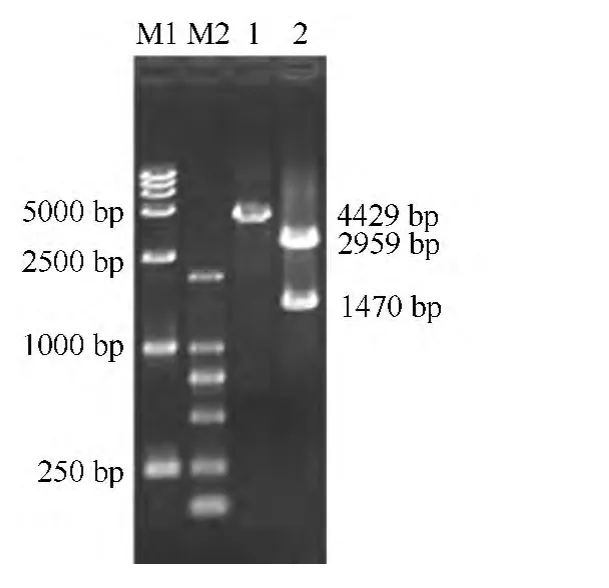
图2 重组质粒pBlue-NDV-NP 的酶切鉴定
由图1可见,RT-PCR 扩增得到NDV LaSota 株NP 基因,1%琼脂糖凝胶电泳中检测结果显示,获得1470 bp 左右产物大小与预计大小相符目的片段。
由图2可见,利用T4 DNA 连接酶将PCR 产物连接载体pBlue,转化到大肠杆菌DH5α,小量提取质粒后进行Xho I 单酶切和Xho I、Eco R I 双酶切鉴定。
以上结果表明,成功构建重组载体pBlue-NDVNP。酶切鉴定正确的重组质粒进行测序验证正确。
2.2 pET28a-NDV-NP 重组表达载体的构建与鉴定详见图3。

图3 重组质粒pET-NDV-NP 的酶切鉴定
由图3可见,限制性内切酶Eco R I 和Xho I 处理pBlue-NDV-NP 和pET-28a 载体,切胶回收,用T4 DNA 连接酶进行连接,转化到感受态DH5α。小量提取质粒,经Xho I 单酶切和Xho I、Eco R I 双酶切鉴定,酶切鉴定与预期结果基本一致,表明成功构建表达载体pET28a-NDV-NP。
2.3 重组表达载体的诱导表达、可溶性分析与目的蛋白纯化 详见图4。

图4 目的蛋白SDS-PAGE
由图4可见,目标载体转化表达工程菌BL21(DE3),终浓度为1 mM/L 的IPTG 诱导5 h 后收集菌液,超声破碎。SDS-PAGE 检测结果表明:pET28a-NDV-NP 上清与沉淀都能在约55 ku 处见到表达条带,目的蛋白与预期大小相符。诱导菌液超声离心处理后取上清液和沉淀进行可溶性分析,结果显示,重组蛋白主要以可溶性形式存在(见图4第1、2 泳道)。通过Ni-NTA 树脂纯化柱纯化NP 重组蛋白,取少量纯化产物进行SDS-PAGE 分析,可见较纯的1 条蛋白条带(见图4第4 泳道)。
2.4 表达蛋白Western-blot 检测 详见图5。

图5 NP 蛋白Western-blot
由图5可见,Western-blot 检测,结果表明,在55 ku 处出现l 条清晰的反应条带,说明重组蛋白在大肠杆菌中得到正确表达并且具有免疫反应原性。
3 小结与讨论
ND 是世界动物卫生组织(OIE)规定的法定上报疫病之一,严重危害着世界各国养禽业的发展[8]。自从该病被发现至今,随着科学技术的发展,对ND 的预控已取得巨大成就。疫苗预防接种是控制ND 的重要措施,研制新型的基因工程疫苗是ND 疫苗研究的重要方向。研究结果表明,在NDV 编码的6 个蛋白质中,F 蛋白和HN 蛋白是NDV 的主要保护性抗原[9]。同时有研究发现,NP蛋白具有很好的免疫原性,单独免疫NP 蛋白就能产生较高的抗NP 蛋白抗体[10]。
原核表达系统具有成本低、表达量高、纯化方便等优点,是获得重组蛋白的首选。大肠杆菌中表达目的蛋白的形式有两种:包含体和可溶性蛋白。本试验将诱导菌液经过超声裂解和表达产物上清与沉淀经蛋白质电泳分析,重组目的蛋白主要存在于超声处理的菌液上清中,便于纯化处理操作。采用蛋白纯化树脂Ni-NTA 柱对目的蛋白进行纯化,不影响蛋白活性、操作简单、纯化效果较好。
本研究通对NDV LaSota 株NP 基因的克隆和原核表达,成功获得重组NDV NP 蛋白。通过其在体外的大量表达,可作为诊断抗原,亦可用其制备抗血清及单克隆抗体,进行疾病诊断,为NDV 在分子水平上进行特异性诊断奠定坚实基础。
另外,NP 基因的克隆与表达,可作为进一步探讨该病毒的分子致病特性及亚单位疫苗的研制提供重要的物质基础。
[1]Y.M.塞夫.禽病学[M].12 版.苏敬良,高 福,索勋,译.北京:中国农业出版社,2012:82-110.
[2]吴艳涛,倪雪霞,万洪全,等.我国部分地区不同动物来源新城疫病毒的分子流行病学研究[J].病毒学报,2002,18(3):264-269.
[3]王忠田,王泽霖,陈溥言,等.新城疫病毒分子生物学最新研究进展[J].动物医学进展,2002,23(2):33-36.
[4]苏长青,卢艳敏,孙焕顷,等.新城疫病毒NP 蛋白的分子生物学特征及其功能[J].江苏农业科学,2009,6:300-301.
[5]Pham H M,Chang K S,Mase M,et al.Characterization of the nucleocapsid protein gene of Newcastle disease virus strains in Japan and development of a restriction enzymebased rapid identifying method[J].Arc Virol,2004,149:1559-1569.
[6]徐彦召,王 青,杭柏林,等.新城疫病毒核衣壳蛋白基因在大肠埃希氏菌中的表达、纯化及其活性分析[J].中国畜牧兽医,2013,40(2):18-22.
[7]萨姆布鲁克J,拉塞尔D W.分子克隆实验指南[M].3版.黄培堂,王嘉玺,朱厚础,等泽.北京:科学出版社,2002:1039-1051.
[8]Miller P J,Decanini E L,Afonso C L.Newcastle disease:evolution of genotypes and the related diagnostic challenges[J].Infect Genet Evol,2010,10(1):26-35.
[9]Karaca K,Sharma J M,Winslow B J,et al.Recombinant fowlpox viruses coexpressing chicken type I IFN and Newcastle disease virus HN and F genes:influence of IFN on protective efficacy and humoral responses of chickens following in ovo or post-hatch administration of recombinant viruses[J].Vaccine,1998,16(6):1496-1503.
[10]Rabu A,Tan W S,Kho C L,et al.Chimeric Newcastle disease virus nucleocapsid with parts of viral hemagglutinin-neuraminidase and fusion proteins[J].Acta Virol.2002,46(4):211-217.
