MR-1通过抑制PERK/Nrf2途径减轻缺氧/复氧诱导的心肌细胞凋亡*
2014-05-16陶天琪王晓礽徐菲菲刘蜜李玉珍刘秀华
陶天琪,王晓礽,徐菲菲,刘蜜,李玉珍,刘秀华
(中国人民解放军总医院病理生理研究室,北京 100853)
·论著·
MR-1通过抑制PERK/Nrf2途径减轻缺氧/复氧诱导的心肌细胞凋亡*
陶天琪,王晓礽,徐菲菲,刘蜜,李玉珍,刘秀华△
(中国人民解放军总医院病理生理研究室,北京 100853)
目的:研究肌原纤维形成调节因子1(MR-1)是否通过抑制蛋白激酶R样内质网激酶(PERK)/核因子E2相关因子2(Nrf2)途径减轻缺氧/复氧(H/R)诱导的心肌细胞凋亡。方法:在原代培养的乳大鼠心肌细胞H/R模型上,采用Annexin V/PI双标法检测心肌细胞的凋亡率;以Western blotting检测葡萄糖调节蛋白78 (GRP78)、磷酸化PERK、Nrf2、活化转录因子4(ATF4)、C/EBP同源蛋白(CHOP)、Bcl-2和Bax的蛋白水平,研究过表达或敲低对于H/R致心肌细胞凋亡的影响及其与PERK/Nrf2途径活化的关系。结果:H/R引起心肌细胞凋亡;过表达MR-1减轻H/R引起的细胞凋亡(P<0.01),下调CHOP表达(P<0.05),引起Bcl-2/Bax值升高(P<0.01),并抑制H/R诱导的PERK磷酸化、Nrf2核转位和ATF4表达(P<0.01)。敲低MR-1加重H/R引起的细胞凋亡(P<0.01)、CHOP表达上调(P<0.05)和Bcl-2/Bax值下降(P<0.01),并加重H/R诱导的PERK磷酸化(P<0.05)、Nrf2核转位和ATF4表达(P<0.01)。结论:MR-1通过抑制PERK/Nrf2途径而减轻缺氧/复氧诱导的心肌细胞凋亡。
细胞凋亡;缺氧/复氧;心肌细胞;肌原纤维形成调节因子1;蛋白激酶R样内质激酶;核因子E2相关因子2
缺血性疾病是严重危害人类健康的常见病[6],尽早恢复组织血供是防治缺血损伤最有效的措施。但缺血再灌注(ischemia/reperfusion,I/R)损伤所致的心肌细胞凋亡、坏死、恶性心律失常和心功能障碍严重影响再灌注疗法疗效和患者预后,是心血管领域亟待解决的重大问题。其中再灌注损伤诱导的心肌细胞凋亡是造成心肌梗死面积扩展、心功能障碍的重要因素,阐明调控心肌细胞凋亡的机制将为缺血再灌注的防治提供新靶点[1]。肌原纤维形成调节因子1(myofibrillogenesis regulator 1,MR-1)是课题组从人类骨骼肌cDNA文库克隆出的人类新功能基因(AF417001),定位于人类染色体2q35,mRNA全长755 bp,编码一段142个氨基酸组成的蛋白质,在心肌、骨骼肌、肾脏和肝脏中高表达[2-4]。课题组前期工作证实,MR-1定位于心肌细胞浆的肌原纤维,缺氧/复氧(hypoxia/reoxygenation,H/R)时心肌细胞MR-1表达下调;过表达MR-1明显减轻H/R导致的肾小管上皮细胞凋亡,提示MR-1具有抗细胞凋亡的作用,其机制尚待阐明。
内质网应激(endoplasmic reticulum stress,ERS)是细胞应激的最初反应,可以整合并启动线粒体和细胞核反应,过度内质网应激诱导细胞凋亡主要通过蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase,PERK)介导的C/EBP同源蛋白(C/EBP homologous protein,CHOP)途径、肌醇需求酶1(inositol-requiring enzyme 1,IRE1)介导的caspase-12[5]和c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)[6]途径所介导。其中PERK通过激活真核细胞起始因子2(eukaryotic initiator factor 2α,eIF2α)上调活化转录因子4(activating transcription factor 4,ATF4),增加CHOP的表达,导致抑凋亡因子Bcl-2蛋白下调和促凋亡因子Bax蛋白上调,引起细胞凋亡[7]。我们既往的工作证实H/R诱导心肌细胞发生严重的ERS反应,表现为内质网应激分子葡萄糖调节蛋白78(glucose-regulated protein 78,GRP78)和PERK表达上调,ERS相关细胞凋亡分子CHOP及其下游分子Bcl-2表达下调,Bax表达上调[8-9],提示PERK途径是介导H/R诱导心肌细胞凋亡的重要信号途径。核因子E2相关因子2(nuclear factor E2-related factor 2,Nrf2)在生理状态下位于细胞浆内的细胞骨架。既往研究证实I/R诱导Nrf2胞核转位,与抗氧化反应元件(antioxidant response elements,AREs)相关靶基因的启动子结合,促使抗氧化蛋白及酶类如NAD(P)H的基因转录增加,减轻氧化反应[10]。最近发现,Nrf2还是PERK的下游底物[6],I/R诱导核转位的Nrf2在细胞核内大量聚集[10],通过作用于ATF4启动子,上调ATF4的转录水平[11],从而正反馈调节PERK途径凋亡相关分子ATF4,提示PERK介导的Nrf2/ATF4相关信号途径可能是I/R诱导细胞凋亡的重要机制。本工作以体外培养的乳大鼠心肌细胞缺氧8 h/复氧16 h模型模拟在体心肌I/R诱导心肌细胞凋亡,分别以腺病毒感染过表达和稳定型小干扰RNA(stealth siRNA,st-RNA)转染技术敲低MR-1表达,研究其对H/ R诱导的心肌细胞凋亡和PERK介导的Nrf2/ATF4信号途径的影响,旨在证实MR-1通过抑制PERK介导的Nrf2/ATF4相关信号途径减轻H/R导致的心肌细胞细胞凋亡,为心肌I/R的防治提供新途径。
材料和方法
1 动物与试剂
清洁级SD乳鼠(出生24 h内)购自北京市军事医学科学院实验动物中心;DMEM培养基购自Gibco;新生牛血清购自杭州四季青生物工程材料研究所;胰蛋白酶、蛋白酶抑制剂和Triton X-100购自Sigma;Annexin V-FITC细胞凋亡检测试剂盒(Annexin V-FITC Apoptosis Direction Kit)购自南京凯基生物工程公司;蛋白电泳分子量(7~175 kD)标记为Bio-Rad产品;蛋白定量试剂盒购自康威世纪公司;脂质体Lipofectamine 2000购自Invitrogen;兔抗人GRP78、PERK、Bcl-2、Bax多克隆抗体、兔抗人甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)单克隆抗体和小鼠抗人CHOP单克隆抗体购自Cell Signaling Technology;兔抗人磷酸化PERK(p-PERK)、ATF4和Nrf2多克隆抗体,Texas red-conjugated驴抗兔抗体,增强化学发光(enhanced chemiluminescence,ECL)试剂盒购自Santa Cruz;辣根过氧化酶标记山羊抗兔和山羊抗小鼠IgG购自Epitomics;胞浆胞核蛋白分离提取试剂盒购自Thermo Fisher Scientific;含DAPI的抗淬灭封片剂购自Vector Laboratories;牛血清白蛋白(bovine serum albumin,BSA)购自Merck;其余化学试剂为国产分析纯产品。
2 乳鼠心肌细胞培养与分组
按我们报道的方法[12]培养乳鼠心肌细胞:出生24 h内乳大鼠常规消毒后无菌操作取出心尖部组织,立即在4℃无菌磷酸盐缓冲液(phosphatebuffered saline,PBS)中洗去残血,分离附着组织后,剪成约1 mm×1 mm×1 mm大小碎块,经0.15%胰蛋白酶37℃反复消化,制备心肌细胞悬液,用含10%新生牛血清的DMEM培养基稀释成细胞密度为3×109/L,接种于75 mm2培养瓶中,置37℃、5% CO2孵箱中进行原代培养。实验前24 h更换无血清DMEM培养基,进行同步化处理后,随机分为以下7 组:(1)正常对照组(control):正常连续培养48 h; (2)缺氧/复氧组(H/R):心肌细胞置于三气培养箱缺氧8 h(37℃,O2含量5%,N2含量90%,CO2含量5%),后置于37℃、5%CO2培养箱复氧培养16 h结束实验;(3)MR-1过表达组(Ad-MR-1):以构建的MR-1重组腺病毒Ad-MR-1感染细胞,使感染复数(multiplicity of infection,MOI;即:加入病毒总数/细胞总数)为50 pfu,感染48 h后结束实验;(4) MR-1敲低组(st-MR-1):针对rat MR-1(NM_ 001134753.1)设计并筛选出25 bp的st-RNA,以50 nmol/L转染细胞,转染方法见下述,转染5 h后更换有血清DMEM培养液;(5)MR-1过表达+缺氧/复氧组(Ad-MR-1+H/R):按照(3)组方法以Ad-MR-1感染细胞20 h后更换无血清DMEM,再按照(2)组程序操作;(6)MR-1敲低+缺氧/复氧组(st-MR-1 +H/R):按照(4)组方法转染st-MR-1,转染后20 h更换无血清DMEM,再按照(2)组程序操作;(7)随机25 bp双链RNA转染对照组(mock):随机合成25 bp且GC含量与st-MR-1一致的双链st-RNA,按照(4)组方法转染操作。
3 腺病毒的包装与感染
按照文献报道方法从质粒pcDNA3.1-hMR-1[13]中扩增目的基因MR-1,委托Invitrogen公司构建包装含有MR-1全长的重组腺病毒Ad-MR-1,结果测定病毒滴度为3.3×1012pfu/L。以该病毒原液感染生长至对数期的细胞,感染前更换有血清的DMEM培养液,每1×105个细胞约加入病毒液10 μL,使感染复数约为50 pfu,感染48 h后结束实验。
4 st-RNA的构建与转染
针对rat MR-1(NM_001134753.1)设计并筛选出25bp的st-RNA(st-MR-1),序列为5’-CGACAGCUAACAAGGCUUCCCAGAA-3’,以50 nmol/L转染细胞,转染方法参照Lipofectamin 2000试剂盒说明书进行,转染5 h后更换有血清DMEM培养液15 h后更换无血清DMEM培养液。
5 Annexin V/PI双标法检测细胞凋亡
培养的乳大鼠心肌细胞以1×104/cm2的密度接种于25 mm2培养皿,参照Annexin V-FITC Apoptosis Direction Kit说明书的方法进行检测:实验处理结束后以含EDTA的0.25%胰酶消化并收集细胞,以0.01 mol/L PBS离心洗涤细胞2次(3 000 r/min离心30 s),洗涤后的沉淀以500 μL Binding Buffer悬浮均匀,分别加入5 μL Annexin V-FITC和5 μL PI,充分混合均匀,室温下避光孵育10 min后,流式细胞术检测早期凋亡率和晚期凋亡率之和。
6 Nrf2细胞荧光染色
细胞以1×104/cm2的密度接种于包被有明胶的盖玻片,实验处理后以PBS洗涤3次,-20℃预冷的冰甲醇预固定5 min,4%多聚甲醛固定15 min,以含有0.1%Triton X-100、含1%BSA的PBS在室温下封闭50 min,以1%BSA配制Ⅰ抗工作液:多克隆兔抗人Nrf2(1∶40),室温下Ⅰ抗工作液孵育1 h,PBS洗涤10 min×3次,以PBS配制Ⅱ抗工作液: Texas red-conjugated驴抗兔(1∶200)。Ⅱ抗工作液于室温下避光孵育1 h,以PBS洗涤10 min×3次,用含DAPI的抗淬灭封片剂封片,激光扫描共聚焦显微镜(Zeiss LSM-510 Meta)观察并采集图像。
7 Nrf2胞浆胞核蛋白分离与蛋白定量
按照胞浆胞核蛋白分离提取试剂盒的操作步骤进行,以BCA法进行蛋白定量。
8 免疫印迹法
取含有80 μg蛋白进行10%聚丙烯酰胺凝胶电泳。电泳后以半干式法电转至硝酸纤维素膜上,0.01mol/L TBST(20 mmol/L Tris-HCl,137 mmol/L NaCl,0.01%Tween-20,pH 7.5)配制的5%BSA室温封闭,加入Ⅰ抗于4℃过夜杂交,不同抗体的稀释度分别为:anti Bcl-2(1∶1 000),anti Bax(1∶1 000),anti CHOP(1∶500),anti ATF4(1∶200),anti Nrf2 (1∶100),antip-PERK(1∶500),antiPERK (1∶1 000),anti GRP78(1∶500),anti GAPDH (1∶1 000);加入辣根过氧化物酶标记的Ⅱ抗(TBST配制,1∶1 000),室温作用2 h后以ECL试剂盒发光显影。以图像分析软件Image-Pro Plus测量各组条带积分吸光度(integrated absorbance,IA;IA=mean absorbance×area)进行半定量分析,并与内参照GAPDH的IA值进行比较。
9 统计学处理
采用SPSS 17.0统计软件分析,数据用均数±标准差(mean±SD)表示,采用单因素方差分析(Oneway ANOVA)进行组间比较,采用q检验进行两两比较;对胞核Nrf2蛋白量与ATF4蛋白量进行相关分析。以P<0.05为差异有统计学意义。
结果
1 MR-1减轻H/R诱导的心肌细胞凋亡
流式细胞术检测细胞凋亡率的结果见图1。缺氧8 h/复氧16 h(H/R组)心肌细胞凋亡率较正常对照组高[(12.1±2.2)%vs(6.3±1.1)%,P<0.01]。与正常对照组比较,重组腺病毒Ad-MR-1感染心肌细胞48 h致MR-1过表达的Ad-MR-1组细胞凋亡率无明显改变(P>0.05);但MR-1过表达明显抑制心肌细胞H/R诱导的心肌细胞凋亡增加[(9.1 ±1.5)%vs(12.1±2.2)%,P<0.01]。以st-RNA敲低MR-1表达后诱导心肌细胞凋亡[(15.9± 2.1)%vs(12.0±1.1)%,P<0.01];与单纯H/R组比较,MR-1敲低加重H/R诱导的心肌细胞凋亡[(17.2±0.8)%vs(12.1±2.2)%,P<0.01]。上述结果提示MR-1具有抑制H/R诱导心肌细胞凋亡的作用。
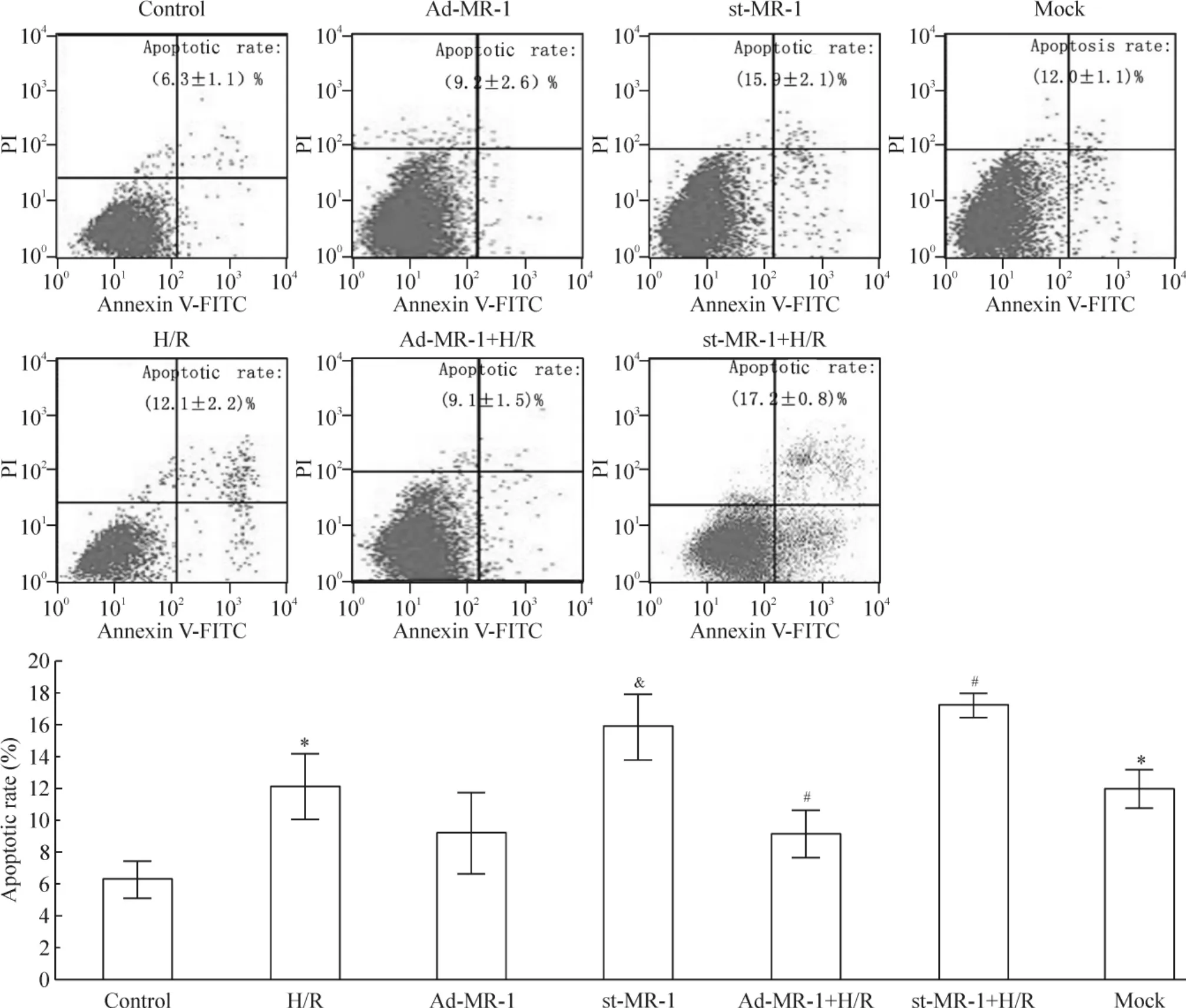
Figure 1.Myofibrillogenesis regulator 1(MR-1)reduced apoptosis induced by hypoxia/reoxygenation(H/R)in vitro as determined by flow cytometric analysis of Annexin V and propidium iodide(PI)double-stained cardiomyocytes.Mean±SD.n=3.*P <0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图1 Annexin V/PI双染的流式细胞术分析MR-1对H/R诱导心肌细胞凋亡的影响
2 MR-1减轻H/R诱导的内质网应激及其相关细胞凋亡
Western blotting检测心肌细胞内质网应激相关分子GRP78的结果见图2。与正常对照组比较,H/ R组GRP78蛋白表达高1.1倍(P<0.01),提示H/R可诱导心肌细胞产生内质网应激。Ad-MR-1组GRP78蛋白表达较正常对照组高1倍(P<0.01),与H/R组无显著差异(P>0.05),但是MR-1过表达减轻了H/R诱导的内质网应激分子GRP78蛋白表达,表现为Ad-MR-1+H/R组GRP78蛋白表达较H/R组低14.2%(P<0.01),提示单纯过表达MR-1可产生内质网应激反应,而MR-1过表达减轻了H/R诱导的内质网应激反应。Mock组GRP78蛋白表达较正常对照组高61.7%(P<0.01),而敲低MR-1(st-MR-1 组)GRP78蛋白表达较Mock组高21.2%(P<0.01);敲低MR-1的心肌细胞在H/R后(st-MR-1+H/R组) GRP78蛋白表达较H/R组高5.6%(P<0.05),提示敲低MR-1本身可以诱导内质网应激反应,并进一步加重H/R诱导的内质网应激反应。

Figure 2.Myofibrillogenesis regulator 1(MR-1)reduced the expression of GRP78 in H/R-treated cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图2 MR-1对H/R心肌细胞GRP78蛋白表达的影响
内质网应激相关细胞凋亡分子CHOP表达的检测结果见图3。与正常对照组比较,H/R组CHOP蛋白表达高16.0%(P<0.01),提示H/R可诱导内质网应激相关细胞凋亡;单纯过表达MR-1对CHOP蛋白表达无明显影响(P>0.05),但是过表达MR-1却减少H/R诱导的CHOP蛋白表达,表现为CHOP蛋白表达较H/R组低9.9%(P<0.05),提示MR-1过表达明显抑制H/R诱导的内质网应激相关细胞凋亡。Mock组心肌细胞CHOP蛋白表达较正常对照组高14.2%(P<0.01);与mock组比较,st-MR-1组心肌细胞CHOP蛋白表达高8.6%(P<0.01);敲低MR-1的心肌细胞行H/R后CHOP蛋白表达较H/R组高5.1%(P<0.05),提示敲低MR-1本身可以诱导内质网应激相关的细胞凋亡,并进一步加重H/R诱导的内质网应激相关的细胞凋亡。

Figure 3.Effect of myofibrillogenesis regulator 1(MR-1)on the expression of ERS-related apoptotic protein CHOP in H/R-treated cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图3 MR-1对H/R心肌细胞CHOP蛋白表达的影响
进一步分析CHOP下游促凋亡因子Bax蛋白和抗凋亡因子Bcl-2蛋白表达的结果见图4。与正常对照组比较,H/R组抑凋亡因子Bcl-2蛋白表达低20.6%,促凋亡因子Bax蛋白表达高18.9%,Bcl-2/ Bax值低33.2%(P<0.01),提示H/R诱导心肌细胞内质网应激相关细胞凋亡的CHOP途径活化。单纯过表达MR-1的Ad-MR-1组Bcl-2和Bax蛋白表达无显著差异(P>0.05),但却明显影响H/R诱导的CHOP下游促凋亡因子Bax蛋白和抗凋亡因子Bcl-2蛋白表达。与H/R组比较,Ad-MR-1+H/R组Bcl-2蛋白表达高29.3%,Bax蛋白表达低15.9%,Bcl-2/Bax值高53.7%(P<0.01),提示MR-1过表达可以减轻H/R诱导的CHOP凋亡途径活化。与正常对照组比较,mock组Bcl-2和Bax蛋白表达分别高23.4%和20.8%,Bcl-2/Bax值低15.2%(P<0.01),st-MR-1组Bcl-2和Bax蛋白表达较mock组分别低41.0%和18.8%(P<0.05),但Bcl-2/Bax值低27.3%(P<0.01);敲低MR-1进一步加重H/ R诱导的Bcl-2下调和Bax上调,表现为与H/R组比较,st-MR-1+H/R组心肌细胞Bcl-2蛋白表达降低25.4%,Bax蛋白表达增高26.2%,Bcl-2/Bax值降低28.8%(P<0.01),提示敲低MR-1本身可以诱导CHOP凋亡途径活化,并进一步加重H/R诱导的CHOP凋亡途径活化。
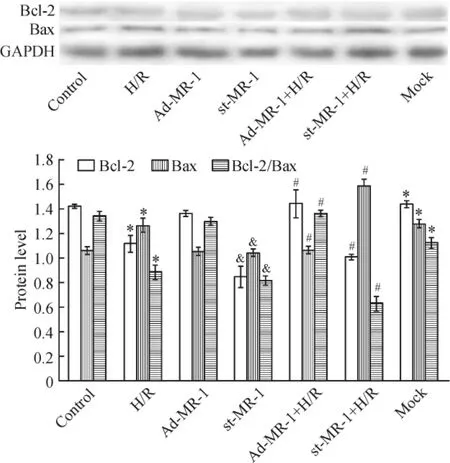
Figure 4.Effect of myofibrillogenesis regulator 1(MR-1)on the expression of anti-apoptotic protein Bcl-2 and proapoptotic protein Bax,and Bcl-2/Bax ratio in H/R-induced cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图4 MR-1对H/R心肌细胞Bcl-2和Bax蛋白表达及Bcl-2/Bax值的影响
3 MR-1通过PERK/Nrf2途径减轻内质网应激相关细胞凋亡
3.1 MR-1抑制H/R诱导的PERK磷酸化Western blotting检测心肌细胞PERK表达与磷酸化的结果见图5。H/R明显上调PERK表达及其磷酸化,其PERK蛋白表达和磷酸化水平分别较正常对照组高24.6%和100%(P<0.01),提示H/R上调PERK蛋白表达和磷酸化,其中磷酸化上调更为明显。单纯过表达MR-1的Ad-MR-1组PERK蛋白表达和磷酸化水平较正常对照组无显著差异(P>0.05);而过表达MR-1后行H/R的心肌细胞,尽管其PERK蛋白表达较H/R组高18.5%(P<0.05),却明显下调PERK磷酸化水平,其PERK磷酸化水平较H/R组低56.5%(P<0.01),提示单纯过表达MR-1对PERK的蛋白表达和磷酸化无明显影响,但明显抑制H/R诱导的PERK磷酸化上调。与正常对照组比较,mock组PERK蛋白表达无显著差异(P>0.05),但其磷酸化水平高22.1%(P<0.01);与mock组比较,单纯敲低MR-1的st-MR-1组PERK蛋白表达和磷酸化水平分别高17.7%和35.0%(P<0.01),而与H/R组比较,敲低MR-1的心肌细胞在H/R后其PERK蛋白表达无明显改变(P>0.05),但PERK磷酸化水平高2.5%(P<0.05),提示敲低MR-1本身可以诱导PERK磷酸化,并进一步上调H/R诱导的PERK磷酸化。

Figure 5.Myofibrillogenesis regulator 1(MR-1)reduced the phosphorylation of PERK in H/R-treated cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图5 MR-1对H/R心肌细胞PERK磷酸化的影响
3.2 MR-1抑制H/R诱导的Nrf2核转位采用细胞免疫荧光染色检测PERK下游分子Nrf2亚细胞分布的结果见图6A。正常对照组Nrf2蛋白主要均匀分布于胞浆,胞核内少量均匀分布。与正常对照组比较,H/R组心肌细胞内Nrf2出现明显的核转位现象,Nrf2主要集聚在细胞核内和核周,且在核内呈团块状分布;单纯过表达MR-1对Nrf2亚细胞分布无明显影响,表现为Ad-MR-1组Nrf2在心肌细胞胞浆内均匀分布,少量均匀分布在核内,与正常对照组类似;但过表达MR-1明显减轻H/R诱导的心肌细胞Nrf2核转位,表现为Ad-MR-1+H/R组心肌细胞内Nrf2均匀分布于胞浆,少量Nrf2均匀分布于核内;与正常对照组比较,mock组Nrf2在胞浆和胞核内均匀分布,但在少数细胞的核内呈团块状分布;与mock组比较,敲低MR-1后心肌细胞Nrf2集中分布在核内和核周,提示敲低MR-1可以造成Nrf2核转位,且敲低MR-1进一步加重H/R诱导的心肌细胞Nrf2核转位,表现为st-MR-1+H/R组心肌细胞内Nrf2团块状积聚于胞核内和核周,极少量分布于胞浆。上述结果表明,H/R造成Nrf2核转位,过表达MR-1明显减轻H/R诱导的Nrf2核转位,敲低MR-1加重H/R诱导的心肌细胞Nrf2核转位。
Western blotting检测Nrf2胞浆与胞核组分蛋白的结果见图6B、C。正常对照组心肌细胞胞浆和胞核中均存在Nrf2蛋白。H/R组Nrf2胞核组分蛋白较正常对照组高47.9%(P<0.01),胞浆组分蛋白无明显改变(P>0.05),提示H/R增加Nrf2蛋白的表达并诱导其核转位。单纯过表达MR-1对心肌细胞Nrf2胞核组分蛋白无明显影响(P>0.05),但胞浆组分蛋白较正常对照组高32.5%(P<0.01);与H/R组比较,过表达MR-1后行H/R的心肌细胞,尽管其胞浆组分蛋白较H/R组高1.2倍,但是明显减少了H/R诱导的Nrf2胞核组分蛋白上调,其Nrf2胞核组分蛋白较H/R低30.6%(P<0.01),提示过表达MR-1增加Nrf2蛋白在胞浆的分布,但抑制H/R诱导的Nrf2核转位。与正常对照组比较,mock组的Nrf2胞核和胞浆组分均升高,表现为胞核组分蛋白高31.4%,胞浆组分蛋白高25.7%(P<0.01);与mock组比较,敲低MR-1仅引起Nrf2胞核组分蛋白上调,表现为Nrf2胞核组分蛋白高18.5%(P<0.01),胞浆组分无显著差异(P>0.05);敲低MR-1进一步加重H/R诱导的心肌细胞Nrf2胞核组分蛋白上调,表现为st-MR-1+H/R组心肌细胞核Nrf2蛋白较H/R组高36.1%(P<0.01),但对胞浆组分无明显影响(P>0.05),提示敲低MR-1进一步上调H/R诱导的Nrf2胞核组分蛋白,加重H/R诱导的Nrf2核转位。
3.3 MR-1抑制H/R诱导的Nrf2下游分子ATF4表达上调Western blotting检测Nrf2的下游分子ATF4表达的结果见图7。与正常对照组比较,H/R 组ATF4蛋白表达高51.6%(P<0.01),提示H/R可通过激活PERK途径导致心肌细胞内质网应激相关凋亡。过表达MR-1对ATF4蛋白表达无明显影响(P>0.05),却明显抑制H/R诱导的ATF4蛋白表达,表现为Ad-MR-1+H/R组ATF4蛋白表达较H/R组低17.7%(P<0.01),提示MR-1过表达通过抑制PERK途径的激活减轻H/R诱导的内质网应激相关细胞凋亡。Mock组的ATF4蛋白表达较正常对照组高19.9%(P<0.01),但st-MR-1组ATF4蛋白表达较mock组高42.4%(P<0.01);敲低MR-1可加重H/R诱导的ATF4蛋白表达,表现为st-MR-1 +H/R组较H/R组高27.4%(P<0.05),提示敲低MR-1本身可以激活PERK途径诱导内质网应激相关细胞凋亡,并通过进一步激活PERK途径加重H/ R诱导的内质网应激相关细胞凋亡。

Figure 6.Myofibrillogenesis regulator 1(MR-1)reduced the nuclear translocation of Nrf2 in H/R-treated cardiomyocytes.A:Nrf2 immunofluorescence assay and DAPI staining under laser scanning confocal microscope (×600);B,C:the levels of nuclear Nrf2 protein and cytoplasmic Nrf2 protein examined by Western blotting.GAPDH was used as a normalization control.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图6 MR-1对H/R心肌细胞Nrf2蛋白核转位的影响
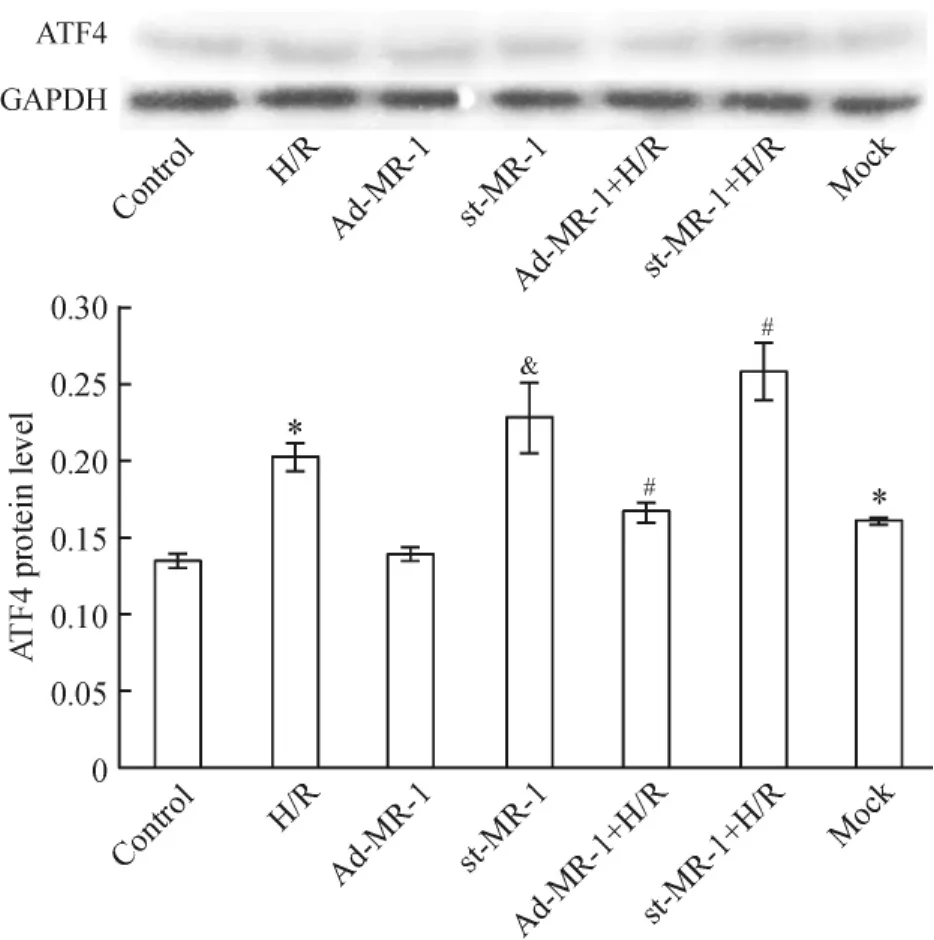
Figure 7.Effect of myofibrillogenesis regulator 1(MR-1)on the expression of ATF4 in H/R-treated cardiomyocytes.Mean±SD.n=3.*P<0.05 vs control;#P<0.05 vs H/R;&P<0.05 vs mock.图7 MR-1对H/R心肌细胞ATF4蛋白表达的影响
3.4 Nrf2胞核组分蛋白与ATF4蛋白表达的相关分析相关分析显示,Nrf2胞核组分蛋白与ATF4蛋白表达呈显著正相关(r=0.935,P<0.01)。
讨论
I/R损伤指缺血一定时间的心肌恢复灌流后,组织损伤反而进行性加重,心肌细胞从可逆损伤转变为不可逆损伤的现象。心肌细胞凋亡和坏死是心肌再灌注损伤的特征之一[14]。减轻心肌细胞凋亡是减轻心肌细胞I/R损伤的关键途径。心肌细胞H/R可在一定程度上模拟心肌缺血再灌注损伤,研究证实H/R可诱导心肌细胞凋亡[8-9]。MR-1是一种从人类骨骼肌cDNA文库克隆出的新功能基因,在心肌、骨骼肌、肾脏和肝脏中高表达[2-4]。本研究首次证实了MR-1通过减轻PERK磷酸化降低H/R诱导的心肌细胞凋亡,对H/R的心肌细胞具有保护作用。
前期工作证实,过表达MR-1明显减轻H/R诱导的肾小管上皮细胞凋亡,提示MR-1可能具有抗细胞凋亡的作用。本研究在新生SD乳鼠心肌细胞H/ R模型上,采用Annexin V/PI双染结合流式细胞术的方法,证实了H/R诱导心肌细胞凋亡,表现为H/ R组细胞凋亡率较正常对照组高,与文献报道一致[15]。MR-1在心肌细胞中有生理性表达[2-4],定位于心肌细胞浆的肌原纤维,H/R时心肌细胞MR-1表达下调。本研究证实MR-1减轻H/R诱导的心肌细胞凋亡。我们以缺氧8 h/复氧16 h处理心肌细胞模拟在体的心肌I/R损伤,发现H/R诱导体外培养的心肌细胞凋亡。我们以含有MR-1全长的重组腺病毒感染心肌细胞,发现过表达MR-1对细胞凋亡率较正常对照组无明显改变,但MR-1过表达明显抑制H/R诱导的心肌细胞凋亡。相反,以st-RNA转染心肌细胞以抑制MR-1的表达,发现敲低MR-1可引起心肌细胞凋亡率的升高,且敲低MR-1加重H/R诱导的心肌细胞凋亡率。
H/R所致的心肌细胞凋亡机制尚未完全阐明。近年来,ERS相关细胞凋亡在心肌细胞凋亡的发病机制中的作用越来越受到重视。ERS是细胞对内外刺激的适应性反应。适度ERS诱导GRP78等内质网伴侣分子表达上调,有利于内质网处理未折叠蛋白或错误折叠蛋白,并促进钙稳态的恢复;持续而严重的ERS可明显上调GRP78的表达,诱导ERS相关的促凋亡因子CHOP的表达及活化,触发ERS相关凋亡途径[6]。CHOP是过度ERS诱导细胞凋亡的关键分子,可直接下调Bcl-2的表达,导致Bax从胞浆转位至线粒体,促进细胞凋亡[16]。Bax和Bcl-2通过形成同源或异源二聚体来调节细胞凋亡。Bax形成同源二聚体时诱导细胞凋亡,Bax与Bcl-2形成异源二聚体时抑制细胞凋亡[17]。Bcl-2与Bax蛋白量的比率决定异二聚体(Bcl-2/Bax)与同二聚体(Bax/ Bax)的比值,故细胞凋亡的发生取决于Bcl-2和Bax的相对浓度,即Bcl-2/Bax值[18]。在正常培养的心肌细胞中,GRP78、CHOP、Bcl-2和Bax均有生理性表达,我们以H/R处理心肌细胞,发现H/R诱导心肌细胞产生ERS相关的细胞凋亡,GRP78、CHOP和Bax表达均高于正常对照组,Bcl-2表达和Bcl-2/Bax值则均低于正常对照组。过表达MR-1可引起适度ERS,但没有引起ERS相关细胞凋亡,表现为GRP78表达较正常对照组高,但对细胞凋亡率及凋亡相关分子的表达无明显影响;过表达MR-1明显减轻H/R诱导的ERS相关心肌细胞凋亡,表现为较H/R组GRP78、CHOP和Bax蛋白表达更低,Bcl-2蛋白表达更高,Bcl-2/Bax值更高;敲低MR-1可产生ERS相关心肌细胞凋亡,表现为较mock组GRP78和CHOP蛋白表达高,Bcl-2/Bax值低;且MR-1敲低后H/R较单纯H/R诱导的ERS相关细胞凋亡更为严重,表现为较H/R组GRP78、CHOP和Bax蛋白表达更高,Bcl-2蛋白表达更低,Bcl-2/Bax比值更低。上述结果提示,MR-1可能作为一个重要的内源性保护分子,在减轻H/R诱导的心肌细胞ERS相关凋亡中发挥重要作用。
PERK是介导ERS相关细胞凋亡的重要感受分子。生理状态下,PERK与内质网相关分子GRP78结合,以无活性复合物的形式存在[19]。未折叠蛋白增多时,GRP78与未折叠蛋白结合而与PERK解离。解离后的PERK发生自身磷酸化而被激活,进一步磷酸化其下游底物eIF2α,选择性上调ATF4。长期过度内质网应激时,ATF4上调CHOP的转录,导致ERS相关细胞凋亡[20]。我们前期研究结果显示,与正常对照组比较,H/R诱导的PERK磷酸化水平、PERK下游信号分子ATF4和CHOP的表达更高,提示H/R诱导PERK介导的ERS相关细胞凋亡[8-9]。Nrf2生理状态时位于细胞浆内的细胞骨架,与其抑制蛋白Keap1结合,处于非游离、非活性状态。当I/ R诱导Nrf2发生核转位,作用于AREs的相关序列,激活相关靶基因,继而启动下游抗氧化蛋白及酶类的基因转录,提高细胞抗氧化应激能力[10]。近期研究证实Nrf2是位于PERK下游的重要转录因子,PERK磷酸化激活Nrf2发生核转位[6],Nrf2的胞核组分蛋白可能作用于ATF4启动子,上调ATF4的转录水平[11],并进一步上调CHOP的转录,导致ERS相关细胞凋亡[20]。根据文献报道,将人视网膜色素上皮细胞敲低Nrf2后行缺氧处理,ATF4的表达较缺氧组明显降低。染色质免疫共沉淀结果显示Nrf2作用于ATF4启动子,且过表达Nrf2激活ATF4启动子,促进ATF4的转录[11]。此外,敲低内皮细胞的Nrf2后行氧化磷脂处理,亦可显著降低ATF4表达[21]。根据文献推断,细胞凋亡可通过内外因素激活PERK/Nrf2核转位/ATF4/CHOP途径发生。
本研究证实,在正常培养的心肌细胞中,Nrf2存在于胞核和胞浆内,但以胞浆内分布为主,PERK、ATF4和CHOP均有生理性表达。我们以H/R处理心肌细胞,发现H/R诱导心肌细胞PERK磷酸化,引起Nrf2核转位,上调ATF4的表达,进而上调CHOP的表达。同时,虽然过表达MR-1对PERK途径无明显影响,表现为PERK磷酸化及下游信号分子的蛋白表达无明显变化,但是过表达MR-1后行H/R处理,可下调H/R诱导的心肌细胞PERK磷酸化,减少Nrf2核转位,下调ATF4和CHOP的蛋白表达,提示过表达MR-1可通过减轻H/R对PERK途径的激活,减轻H/R诱导的ERS相关细胞凋亡。此外,敲低MR-1可诱导PERK磷酸化介导的ERS相关细胞凋亡,表现为PERK磷酸化高于MOCK组,Nrf2核转位较mock组更明显,ATF4和CHOP蛋白表达较mock组高;敲低MR-1后行H/R处理可通过进一步激活H/R诱导的PERK途径,进而加重H/R诱导的ERS相关细胞凋亡,表现为PERK磷酸化较H/R组高,Nrf2核转位较H/R组更明显,ATF4和CHOP蛋白表达较H/R组高。在此基础上对ATF4蛋白表达和胞核Nrf2蛋白之间进行相关分析发现,ATF4蛋白表达与Nrf2胞核组分蛋白呈正相关,提示Nrf2核转位介导的凋亡信号途径可能通过促进ATF4蛋白表达引起细胞凋亡。
另有文献报道,转录因子Nrf2和ATF4具有相似的靶基因,存在协同和拮抗作用。两者可直接诱导AREs相关基因的转录[22],但也可直接作用于CHOP启动子,ATF4促进CHOP转录,Nrf2抑制CHOP转录[23]。此外,Nrf2也可通过解除ATF4与CHOP的结合,进而抑制CHOP转录[24]。根据研究结果推断,我们认为在H/R引起心肌细胞PERK介导的ERS相关细胞凋亡的过程中,Nrf2发生核转位,促进ATF4的表达。虽然Nrf2可直接或间接下调CHOP的转录,但是ATF4上调促进CHOP的表达增加起主要作用。这提示H/R通过PERK介导的Nrf2核转位/ATF4/CHOP途径诱导心肌细胞凋亡,而MR-1通过抑制上述途径减轻H/R诱导的心肌细胞凋亡。
综上所述,我们认为MR-1通过抑制过度ERS发挥减轻心肌H/R损伤的作用,表现为降低H/R诱导的GRP78蛋白表达,抑制PERK介导的Nrf2核转位/ATF4/CHOP途径,减轻H/R诱导的ERS相关细胞凋亡。但MR-1对在体大鼠心肌I/R损伤的心肌保护作用尚待进一步研究。
[1]刘秀华,唐朝枢.缺血再灌注损伤的防治——从实验室到临床[J].中华心血管病杂志,2006,34(8):677-679.
[2]Li TB,Liu XH,Feng S,et al.Characterization of MR-1,a novel myofibrillogenesis regulator in human muscle[J].Acta Biochim Biophys Sin,2004,36(6):412-418.
[3]徐菲菲,刘秀华,王彦珍,等.肌原纤维调节因子-1在心肌肥大中的作用研究[J].中国病理生理杂志,2006,22(3):443-447.
[4]王晓礽,刘秀华,宋泉声,等.人肌纤生成调节因子-1抗体的制备、鉴定及其在乳鼠心肌细胞中的应用[J].中国病理生理杂志,2010,26(1):42-47.
[5]Kim EM,Shin EJ,Choi JH,et al.Matrix metalloproteinase-3 is increased and participates in neuronal apoptoticsignaling downstream of caspase-12 during endoplasmic reticulum stress[J].J Biol Chem,2010,285(22):16444-16452.
[6]Hetz C.The unfolded protein response:controlling cell fate decisions under ER stress and beyond[J].Nat Rev Mol Cell Biol,2012,13(2):89-102.
[7]Pahl HL.Signal transduction from the endoplasmic reticulum to the cell nucleus[J].Physiol Rev,1999,79(3): 683-701.
[8]徐菲菲.钙网蛋白介导的内质网应激相关凋亡参与心肌细胞缺氧/复氧损伤[D].北京:中国人民解放军军医进修学院,2009.
[9]王琛,李玉珍,王晓礽,等.西洋参茎叶总皂苷通过抑制过度内质网应激减轻大鼠心肌细胞缺氧/复氧损伤[J].中国病理生理杂志,2012,28(1):22-28.
[10]Leonard MO,Kieran NE,Howell K,et al.Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury[J].FASEB J,2006,20(14): 2624-2626.
[11]Miyamoto N,Izumi H,Miyamoto R,et al.Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE-19/HPV-16 cells[J].Invest Ophthalmol Vis Sci,2011,52(3):1226-1234.
[12]Liu XH,Wu XD,Cai LR,et al.Hypoxic preconditioning of cardiomyocytes and cardioprotection:phosphorylation of HIF-1α induced by p42/p44 mitogen-activated protein kinases is involved[J].Pathophysiology,2003,9(4):201-205.
[13]Wang X,Liu X,Wang S,et al.Myofibrillogenesis regulator 1 induces hypertrophy by promoting sarcomere organization in neonatal rat cardiomyocytes[J].Hypertens Res,2012,35(6):597-603.
[14]Fliss H,Gattinger D.Apoptosis in ischemic and reperfused rat myocardium[J].Circ Res,1996,79(5):949-956.
[15]张峰,梅其芮,张涛,等.缺氧预适应抑制缺氧/复氧诱导心肌细胞凋亡的作用及机制[J].中国病理生理杂志,2005,21(2):351-355.
[16]McCullough KD,MartindaleJL,KlotzLO,etal.Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl-2 and perturbing the cellular redox state[J].Mol Cell Biol,2001,21(4):1249-1259.
[17]Lalier L,Cartron PF,Juin P,et al.Bax activation and mitochondrial insertion during apoptosis[J].Apoptosis,2007,12(5):887-896.
[18]王卫东.Bcl-2/Bax比率与细胞“命运”[J].中国肿瘤生物治疗杂志,2007,14(4):393-396.
[19]Bertolotti A,Zhang Y,Hendershot LM,et al.Dynamic interaction of BiP and ER stress transducers in the unfoldedprotein response[J].Nat Cell Biol,2000,2(6):326-332.
[20]Endo M,Oyadomari S,Suga M,et al.The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice[J].J Biochem,2005,138(4):501-507.
[21]Afonyushkin T,Oskolkova OV,Philippova M,et al.Oxidized phospholipids regulate expression of ATF4and VEGF in endothelial cells via NRF2-dependent mechanism:novel point of convergence between electrophilic and unfolded protein stress pathways[J].Arterioscler Thromb Vasc Biol,2010,30(5):1007-1013.
[22]He CH,Gong P,Hu B,et al.Identification of activating transcription factor 4(ATF4)as an Nrf2-interacting protein.Implication for heme oxygenase-1 gene regulation [J].J Biol Chem,2001,276(24):20858-20865.
[23]Cullinan SB,Diehl JA.PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress[J].J Biol Chem,2004,279(19):20108-20117.
[24]Zong ZH,Du ZX,Li N,et al.Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells[J].Biochim Biophys Acta,2012,1823(8):1395-1404.
Myofibrillogenesis regulator 1 attenuates hypoxia/reoxygenation-induced apoptosis of cardiomyocytes by inhibiting PERK/Nrf2 pathway
TAO Tian-qi,WANG Xiao-reng,XU Fei-fei,LIU Mi,LI Yu-zhen,LIU Xiu-hua
(Department of Pathophysiology,Chinese PLA General Hospital,Beijing 100853,China.E-mail:xiuhualiu98@163.com)
AIM:To investigate the effect of myofibrillogenesis regulator 1(MR-1)on hypoxia/reoxygenation (H/R)-induced apoptosis of cardiomyocytes and to study the role of protein kinase R-like endoplasmic reticulum kinase (PERK)/nuclear factor E2-related factor 2(Nrf2)pathway.METHODS:In the H/R model of primarily cultured neonatal rat cardiomyocytes,the apoptosis was assessed by Annexin V/PI double staining.Western blotting was used to detect the protein levels of glucose-regulated protein 78(GRP78),phosphorylated PERK,Nrf2,activating transcription factor 4 (ATF4),C/EBP homologous protein(CHOP),Bcl-2 and Bax.The effects of over-expression or knockdown of MR-1 on the apoptosis and the PERK/Nrf2 pathway were determined.RESULTS:H/R induced the apoptosis of cardiomyocytes.Compared with H/R group,MR-1 over-expression attenuated H/R-induced apoptosis(P<0.01),down-regulated CHOP expression(P<0.05),and increased Bcl-2/Bax ratio(P<0.01).MR-1 over-expression suppressed H/R-induced PERK phosphorylation,Nrf2 nuclear translocation and ATF4 expression(P<0.01).However,MR-1 knockdown aggravated H/R-induced apoptosis(P<0.01),up-regulated CHOP expression(P<0.05),and decreased Bcl-2/Bax ratio(P<0.01).MR-1 knockdown up-regulated H/R-induced PERK phosphorylation(P<0.05),Nrf2 nuclear translocation and ATF4 expression(P<0.01).CONCLUSION:MR-1 suppresses H/R-induced cardiomyocyte apoptosis by inhibiting PERK/Nrf2 pathway.
Apoptosis;Hypoxia/reoxygenation;Cardiomyocytes;Myofibrillogenesis regulator 1;Protein kinase R-like endoplasmic reticulum kinase;Nuclear factor E2-related factor 2
R363.1
A
10.3969/j.issn.1000-4718.2014.02.001
1000-4718(2014)02-0193-10
2013-10-15
2014-01-02
国家自然科学基金资助项目(No.81170140;No.81070130)
△通讯作者Tel:010-66939763;E-mail:xiuhualiu98@163.com
