利用谱图校正提高色谱-质谱联用的分析效率
2014-05-08陆文渊沈诚频殷薛飞刘晓慧杨芃原
陆文渊, 张 扬, 沈诚频, 殷薛飞, 刘晓慧, 杨芃原
(1.复旦大学化学系,上海200433;2.复旦大学上海医学院生物医学研究院,上海200032)
色谱技术作为日渐成熟的蛋白质组学研究中最为重要的分离手段之一,与样品预处理以及生物信息学分析一样,存在着蛋白质组学研究中一些需要解决的难题。色谱条件的选择直接影响蛋白质的分离效率与质量,并间接地对实验结果产生着决定性的影响。但是,迄今为止的很长一段时间,无论是蛋白质的定性还是定量,大多数实验分析都以质谱数据为主,而大量色谱实验数据普遍被忽视。
色谱数据理应被重视和运用于蛋白质的分析中,但在实际分析中,色谱实验数据又存在着难以处理的问题,如几乎所有样品的色谱数据中都会出现保留时间的偏移,这种偏移与液相流动速率、温度以及柱压等因素有关[1];此外,样品浓度的不同不仅会使色谱离子流图的响应值发生变化,还会使得特征峰发生保留时间上的偏移。因此,建立理想的校正方法,可以将不同浓度样品的特征峰对齐,从而有助于在低浓度谱图中鉴定出更多的肽段。
事实上,即使是同浓度的同种样品在连续进行的重复实验中,色谱保留时间的偏移仍无法消除。对于复杂样品来说,大量酶解肽段无一例外地出现了保留时间偏移,其结果势必会对鉴定造成无法估计的影响。因此,对大部分流出峰进行校正同样也可以提高复杂体系中的肽段与蛋白质鉴定数量。
另外,除了色谱保留时间的偏移,质谱m/z信号也存在无法避免的质量偏移[2],对蛋白质鉴定造成影响。因此,对于蛋白质定性定量分析实验,保留时间和m/z信号双方面的校正是一个非常有意义且富有挑战的尝试。本文以复杂样品的定性分析为例,提出谱图校正与后续处理流程,并且得到了较好的实际分析结果。
1 实验部分
1.1 仪器与试剂
BreezeTMHPLC色谱仪,购自美国Waters公司;EASY-nLC 1000纳升级液相色谱、Heraeus Megafuge 1.0R通用离心机,购自美国 ThermoFisher公司;TripleTOF5600质谱仪,购自美国 ABSciex公司;Concentrator plus真空离心浓缩仪,购自美国Eppendorf公司;Symmetry C18反相色谱柱(250 mm×4.6 mm,5 μm,10 nm),购自美国Waters公司;AQUASIL C18 反相色谱柱(50 cm ×75 μm,2 μm),购自美国ThermoFisher公司;乙腈(ACN)、氨水(NH3·H2O),色谱纯,购自德国Merck公司;胞内蛋白酶(lys-C)、胰蛋白酶(trypsin),纯度不低于95%;三氟乙酸(TFA)、二硫苏糖醇(DTT)、吲哚乙酸(IAA)、碳酸氢铵均购自美国Sigma-Aldrich公司,使用前稀释至指定浓度。
1.2 实验样品
样品1:全细胞提取的人脐静脉内皮细胞(HUVEC),用二甲基亚砜(DMSO)(溶解于8 mol/L尿素、100 mmol/L氯化钠溶液中)刺激,共330 μg;样品2:全细胞提取的HUVEC,用NEDD8激活酶抑制剂(MLN4924,MLN,溶解于8 mol/L尿素、100 mmol/L氯化钠溶液中)刺激,共330 μg;样品3:QconCAT蛋白质混合物,蛋白质绝对量跨度为4个数量级(见表1),共26 μL。以上样品皆由中国人类蛋白质计划(CNHPP)提供。
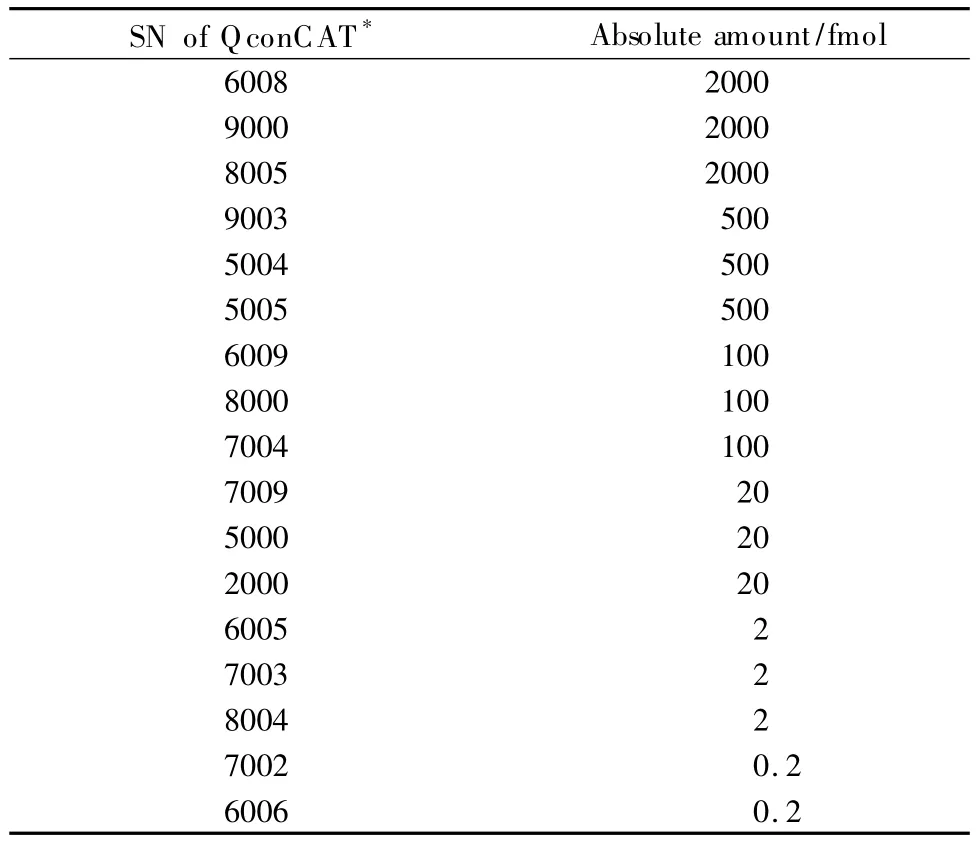
表1 QconCAT蛋白质混合物绝对浓度表Table 1 Absolute amount of QconCAT protein mixture
1.3 样品处理
对3份样品分别进行还原烷基化处理,具体方法:在56℃下,用10 mmol/L的DTT反应0.5 h,随后在37℃下,用20 mmol/L的IAA反应0.5 h。
对刺激的两份HUVEC样品(即样品1和样品2)进行以下酶解:先在37℃下,用胞内蛋白酶(酶∶蛋白质 =1∶50,质量比,下同)酶解 3 h,用 50 mmol/L的碳酸氢铵稀释至两倍体积,再在37℃下,依次用胰蛋白酶(酶∶蛋白质=1∶50)和胰蛋白酶(酶∶蛋白质 =1∶100)酶解8 h和6 h;对 Qcon-CAT蛋白质混合物(即样品3)进行以下酶解:在37℃下,用100 ng胞内蛋白酶酶解3 h,再依次用胰蛋白酶100 ng和50 ng分别酶解8 h和6 h;再加入三氟乙酸至其质量分数为0.1%来终止酶解,并用C18反相柱除盐;将QconCAT酶解溶液平均分为两份,分别加入DMSO与MLN刺激的细胞酶解液中,混合均匀后真空干燥,并复溶于高pH的色谱流动相A相溶液中,两份分别标记为样品Ⅰ和样品Ⅱ。
1.4 色谱-质谱联用分析
1.4.1 第一维高pH反相色谱流程
选用 Symmetry C18色谱柱(250 mm×4.6 mm,5 μm,10 nm)。流动相 A 相为乙腈-水(2∶98,v/v)(用氨水调pH等于10.0),B相为乙腈-水(98∶2,v/v)(用氨水调pH等于10.0)。梯度洗脱程序如下:初始B相为5%,在5 min内升至8%,在35 min内从8%升至18%,在22 min内从18%升至32%,在2 min内从32%陡增至95%并保持4 min,最后在4 min内从95%降至5%。将Ⅰ和Ⅱ两份样品分别按时间分布收集65个馏分(每1 min收集一个馏分),冻干,之后再按顺序每11份(最后一组为10份)合并成6个馏分准备进行第二维分离。
1.4.2 第二维反相色谱分离
选用 Aquasil C18色谱柱(50 cm ×75 μm,2 μm)。流动相A相为2%的乙腈与98%的水(含0.1%TFA),B相为98%的乙腈与2%的水(含0.1%TFA)。梯度洗脱程序如下:初始B相为0,在30 min内从0升至10%,在210 min内从10%升至20%,在100 min内从20%升至35%,在5 min内从35%升至80%,在80%保持10 min,最后在5 min内从80%降至0,并且保持40 min。
1.4.3 质谱扫描
喷针外加电压选用2.3 kV;扫描范围为m/z 350~1 500的全扫描,扫描方式为高分辨模式(high resolution);串级质谱扫描范围为m/z 100~1 250、扫描模式为信息依赖性获取模式(IDA,information dependent acquisition)。
2 结果与讨论
2.1 色谱校正方案选择
2.1.1 特征峰检测与谱图校正的次序先后研究
谱图校正的目标是在信号噪声中,将不同实验中存在保留时间偏移、但具有相同特征的肽段流出峰对齐。自1998年以来,谱图校正对齐的研究报道已有过不少,Nielsen等[3]首先建立了一套相关优化规整算法(COW,correlated optimized warping algorithm),随后Wang等[4]在此基础上研究了动态时间规整算法(DTW,dynamic time warping algorithm),此外,还有如 May 等[5]编写的 msInspect、Li等[6]与 Fugmann 等[7]编写的 SpecArray 以及 Katajamaa等[8]编写的MZmine等软件可以进行谱图校正对齐。
概括起来,可以把这些校正方法归为两类。第一类方法是在特征峰检测前对原始数据进行校正对齐,这类方法通过计算理想曲线方程将一次实验的色谱保留时间对齐至另一次实验。然而,曲线方程将整个色谱中的保留时间差异一齐纳入了计算,因此,该方法并不能保证将单个肽段进行校正对齐。第二类方法则是利用检测完的特征峰列表进行校正对齐,并且允许个别肽段存在一定范围的保留时间差异。但是,这类方法依赖于对特征峰的预先检测,而且无法利用原始谱图数据的信息,因此特征峰检测出现的错误会直接影响到整个谱图的校正与对齐。总之,这两类方法均只能用于相似度比较高的数据之间,而对于不同的样品很有可能会出现错误,比如比较正常和癌变组织蛋白质的实验中。
由于第二类方法需要事先进行特征峰的检测,会在谱图校正前引入额外的影响因素,因此,我们在第一类方法中选择合适的算法并做进一步研究。研究的内容主要是从第一类方法中筛选出合适的算法,并挑选和利用现有的软件进行改进,以符合预期的校正功能。
2.1.2 校正算法的研究
上文提到的第一类方法,主要有秩最小化规整[9]、抽象子空间差异规整[10]、动态时间规整[11]、参数时间规整[12]以及相关性优化规整等。一直以来,这些方法只是在一般有机物的色谱鉴定中使用,无法直接运用于蛋白质组学的实验中。
当比较两次LC/MS实验谱图时,Fischer等[13]并没有直接将两次谱图发生的保留时间偏移进行校正对齐,而是选择以其中一次实验的保留时间作为函数的自变量,将两次实验谱图的保留时间差值作为因变量,再进行校正处理。由于计算量过于庞大,Fischer等开始并没有将所有的数据进行校正,只是选取了一些可以得到鉴定的肽段流出峰。随后,该方法被广泛用于色谱-质谱联用数据的校正[14,15],并且为后续的定性定量处理提供帮助。
2008年以来,Christin等[16]认为基于总离子流(TIC)图的保留时间校正方法只运用了保留时间一维信息,并没有考虑到LC-MS数据中不同m/z信号对总离子流图的贡献,他们认为用于LC-MS数据时间漂移校正的方法应该对每一个有贡献的m/z响应进行校正。然而,对于特定的一个色谱流出时间点,其对应的m/z响应信号有很多个,而其中真正的目标分析物信号却只占小部分,因此,在运用校正方法之前,必须对这些质谱信号进行筛选,选择出高质量的质谱信号后再进行校正。为此,“组分检测算法”[17]的变量选择方法被运用到对质谱数据的筛选中。该算法具有计算时间快,对色谱峰形影响小等特点,与色谱领域所运用的 DTW、PTW 和COW算法相结合,可广泛用于蛋白质组学LC-MS的时间漂移校正。
结合前文总结的两类校正方法的特点,本文最终选用Progenesis LC-MS软件的算法,筛选出大量不同的色谱峰,并对所有色谱峰进行校正,之后,从对齐的多次实验色谱图中再次检测特征峰,并生成可用于搜库的.mgf文件。通过Mascot软件完成搜库后,再将获得的.xml文件导入Progenesis LC-MS软件,进行后续的分析处理。
2.1.3 其他谱图校正方法
除了上文提到的以及我们最终选用的方法外,Aebersold 等[18]与 Escher等[19]采用 iRT(i-rentention time)的方法进行了校正。他们选取了一系列被称为iRT肽段的标准肽段(见表2)作为参照加入实验样品中,以此对样品肽段流出峰进行校正。对于在两个iRT肽段间流出的肽段x,其iRT值计算方法为:iRTx=[(RTx-RT1)/(RT2-RT1)]×100。

表2 iR T肽段的序列与对应iR T值Table 2 iR T-peptide sequences and corresponding iR T values
同一条肽段在不同实验中经过换算后得到的iRT值是相同的,那么,只要选定一次实验的保留时间计算得到该肽段的iRT值,其他实验中的该肽段流出峰保留时间就可以匹配上去,最终达到校正和对齐的效果。但是,由于11条iRT肽段将色谱图分割成了10个区间,导致谱图的校正对齐必须在分割后的区域内分别完成,因此,我们最终并没有选择该方法。事实上,iRT的方法在很多时候都被用于保留时间的预测以及质谱多反应检测(MRM)中。
2.2 利用Progenesis LC-MS软件的校正结果
2.2.1 预校正结果的分析
本文中,我们首先对样品Ⅰ和样品Ⅱ的各6个馏分分别进行第二维反相色谱分离,再进入质谱,连续进行三次串级扫描,共计得到36个.wiff文件,总大小约400 G。进而,我们选取样品Ⅰ的第四馏分进行了预校正分析。
对于任何一次HPLC-MS/MS实验数据,都可以得到一系列可视化图(如图1)。图1a的横坐标与纵坐标分别为m/z值与保留时间,图中点的颜色深浅代表对应的m/z与保留时间的色谱峰强度;图1b为最常见的总离子流图。我们将样品Ⅰ第四馏分的3次重复进样结果作了校正计算,图2为样品Ⅰ第四馏分的第二次与第三次重复进样谱图校正前后的情况。从图中可以看出,几乎所有的流出峰都得到了比较良好的校正与对齐(见图2b)。
图1 样品Ⅰ第四馏分第二次进样的(a)离子强度图与(b)总离子流图Fig.1 (a)Ion intensity map and(b)total ion chromatogram of the 2nd injection for the 4th fraction of sampleⅠ

图2 样品Ⅰ第四馏分第二次进样与第三次进样(a)校正前与(b)校正后的总离子流图比较Fig.3 Comparison of total ion chromatograms of the 2nd and the 3rd injections for the 4th fraction of sampleⅠ(a)before alignment and(b)after alignment
将3次重复实验的谱图进行校正对齐后,我们分别计算了它们之间的谱图相似率(见表3)。由预校正相似率结果可知,在谱图校正方面,我们采用的策略是可行的。因此,我们将该方法运用到了两个样品(Ⅰ和Ⅱ)总共12个馏分的36次实验数据的批量处理中。
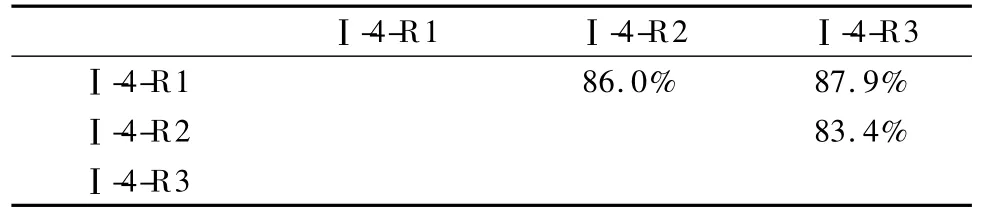
表3 样品Ⅰ第四馏分3次重复进样之间的谱图相似率Table 3 Similarities among three repeat injections of the 4th fraction in sampleⅠ
2.2.2 通过QconCAT标准蛋白质检验谱图校正
在样品处理过程中,我们在两种不同药物刺激的HUVEC样品中等量加入了QconCAT标准蛋白质。因此,可以通过对QconCAT标准蛋白质的鉴定来考察谱图校正的情况。
将校正后的谱图文件导出为.mgf文件,然后,使用Mascot软件进行MS/MS搜索,使用QconCAT标准蛋白库,肽段误差允许为10 ppm,串级质谱误差允许为1 Da。将搜库结果生成.xml文件,并导入Progenesis LC/MS软件,该软件将自动参考谱图搜索结果把谱图文件中的已鉴定肽段流出峰挑选出并生成列表。
最后,我们将样品Ⅰ和Ⅱ各6个馏分的处理结果按照不同的重复次数进行合并,得到了样品Ⅰ及样品Ⅱ总鉴定QconCAT标准蛋白质数的3次重复文件,分别标记为Ⅰ-R1、Ⅰ-R2、Ⅰ-R3、Ⅱ-R1、Ⅱ-R2和Ⅱ-R3。对于这6个新生成的文件,我们将它们各自鉴定到的QconCAT标准蛋白质作图进行两两比较,对作比较的两个文件中鉴定到的标准蛋白质的保留时间进行线性拟合处理,得到相关系数(R)。
通过相关系数的“热图”可以直观地发现:两种不同药物刺激的样品中,鉴定到的QconCAT标准蛋白质的相关系数在0.9附近,而同一样品的重复进样之间相关系数都在0.98以上(见图3)。推测造成这种结果的原因,主要为DMSO和MLN两种不同的刺激使得HUVEC细胞内的蛋白质发生了变化,而在不同的复杂体系背景下,蛋白质的酶解肽段之间会发生互相干扰,影响到保留时间甚至是峰强度。因此,依靠谱图校正的方法,我们在今后的研究中将会把肽段互相影响的因素作为研究方向。

图3 样品Ⅰ和Ⅱ所有重复实验中QconCAT标准蛋白质鉴定的相关系数的热图Fig.3 Thermograph for correlation coefficients of identified QconCAT standard proteins among three repeat injections of sampleⅠandⅡ
2.2.3 HUVEC细胞中蛋白质的定性、定量结果比较
最后,我们将校正前后的实验数据在Mascot软件中重新进行了搜库,使用Swissprot标准蛋白库,肽段误差允许为10 ppm,串级质谱误差允许为1 Da。对未进行谱图校正而直接进行搜库的结果统计处理后,显示从DMSO处理的HUVEC细胞中鉴定到了6 535个蛋白质,从MLN处理的HUVEC细胞中鉴定到了6 925个蛋白质。
经过谱图校正之后,DMSO和MLN处理的HUVEC细胞分别鉴定到了8 089个和8 094个蛋白质(见图4)。蛋白质的鉴定数量平均提高了近1 000个左右,说明我们的校正方法可以提高复杂体系中蛋白质的鉴定率。
此外,我们还对DMSO和MLN刺激的HUVEC细胞内蛋白质进行了依靠谱图计数的相对定量分析[20,21],在 DMSO 刺激的 HUVEC 细胞中得到了6 308个蛋白质,而MLN刺激的细胞中得到了6 873个蛋白质,我们以两倍比值差异进行了两个组分的差异分析,相比于DMSO刺激的细胞,MLN刺激的HUVEC细胞中有930个蛋白质上调,274个蛋白质下调(见图5)。
2.3 存在的问题
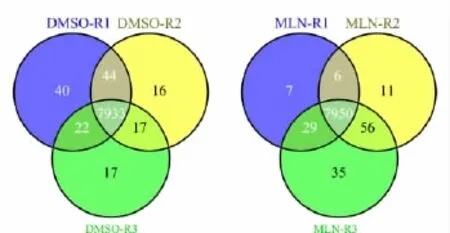
图4 谱图经校正后的DMSO与MLN刺激的HUVEC细胞内蛋白质鉴定结果Fig.4 Protein identification results in HUVEC treated with DMSO and MLN after spectrum alignment

图5 DMSO和MLN刺激导致的HUVEC细胞内蛋白质的差异Fig.5 Differential expression of proteins in HUVEC treated with DMSO or MLN
该方法的问题主要存在于低质荷比范围发生的质量偏移。在预校正分析的样品Ⅰ的两次重复中,我们把谱峰校正、合并后,放大低质荷比端,结果发现:两次实验的质荷比无法完全重现(见图6上图),而中等质荷比端则没有发现此问题(见图6下图)。目前解决该问题的方法是进行人工修正,暂时还没有软件自动校正的方法,这也是下一步需要研究关注的内容。
3 结论
本文通过对不同刺激的HUVEC细胞内蛋白质进行分析,发展并检验了一种谱图校正流程。该流程首先对复杂蛋白质样品进行甲基化和双酶解,并加入QconCAT标准蛋白质作为参照,然后通过高效液相色谱-串级质谱进行分离鉴定,最后通过Progenesis LC/MS软件和Mascot软件对实验数据进行校正和分析。谱图相似度的比较及对QconCAT标准蛋白质参照物的鉴定结果都证明了该流程比起其他方法更加快捷而有效,并且拥有高通量高灵敏度的优点。

图6 低质荷比区域(上图)与中等质荷比区域(下图)的离子强度图Fig.6 Ion intensity maps of low m/z zone(up)and medium m/z zone(down)
[1] Wang P,Tang H,Fitzgibbon M P,et al.Biostatistics,2007,8(2):357
[2] Kim S,Mischerikow N,Bandeira N,et al.Mol Cell Proteomics,2010,9(12):2840
[3] Nielsen N P V,Carstensen J M,Smedsgaard J.J Chromatogr A,1998,805(1):17
[4] Wang S,Yao J,Liu J,et al.Medical Physics,2009,36(12):5595
[5] May D,Law W,Fitzgibbon M,et al.J Proteome Res,2009,8(6):3212
[6] Li X J,Yi E C,Kemp C J,et al.Mol Cell Proteomics,2005,4(9):1328
[7] Fugmann T,Neri D,Roesli C.Proteomics,2010,10(14):2631
[8] Katajamaa M,Oresic M.BMC Bioinformatics,2005,6(1):179
[9] Fraga C G,Prazen B J,Synovec R E.Anal Chem,2001,73(24):5833
[10] Yu Y J,Wu H L,Fu H Y,et al.J Chromatogr A,2013,1302:72
[11] Ramaker H J,van Sprang E N M,Westerhuis J A,et al.Anal Chim Acta,2003,498(1/2):133
[12] Eilers P H C.Anal Chem,2004,76(2):404
[13] Fischer B,Grossmann J,Roth V,et al.Bioinformatics,2006,22(14):e132
[14] Tsai T H,Tadesse M G,Di Poto C,et al.Bioinformatics,2013,29(21):2774
[15] Lin H,He L,Ma B.Bioinformatics,2013,29(14):1768
[16] Christin C,Smilde A K,Hoefsloot H C J,et al.Anal Chem,2008,80(18):7012
[17] Christin C,Hoefsloot H C J,Smilde A K,et al.J Proteome Res,2010,9(3):1483
[18] Rost H,Malmstrom L,Aebersold R.Mol Cell Proteomics,2012,11(8):540
[19] Escher C,Reiter L,MacLean B,et al.Proteomics,2012,12(8):1111
[20] Old W M,Meyer-Arendt K,Aveline-Wolf L,et al.Mol Cell Proteomics,2005,4(10):1487
[21] Asara J M,Christofk H R,Freimark L M,et al.Proteomics,2008,8(5):994
