超高效液相色谱-串联质谱法检测保健食品中激素类物质
2014-05-08张晓光郑雅梅
张晓光,郑雅梅,李 强,杨 刚,张 岩
(1.河北省食品检验研究院,河北 石家庄 050091;2.河北医科大学,河北 石家庄 050017)
近年来,随着人们生活水平的不断提高,对保健食品的需求量逐年增大,不法商贩为了追求更高利益,向保健品中违法添加西药类物质屡见不鲜。糖皮质激素具有广泛的临床用途,因而成为不法商家在保健品中非法添加的目标物质,如果误食了非法添加含有糖皮质激素类物质的保健食品,可能会导致诸多不良反应,造成人体机体代谢紊乱、发育异常或潜在致畸致癌风险[1-2]。由此可见,加强对保健品中非法添加的监管,建立保健品中激素类检测方法十分必要。
精确先进的激素测定方法是实现激素残留有效监督的重要手段,目前,激素类物质的检测方法主要有高效液相色谱法[3-5]、毛细管色谱法[6]、薄层色谱法[7]、气相色谱-质谱联用法[8]、液相色谱-质谱联用法[9-11]等。本文介绍了一种超声萃取-超高效液相色谱-质谱联用检测方法能快速分析检测保健食品中10种糖皮质类类西药成分,该方法主要的优势在于检测时间短,方法准确、简便快速,能够为保健品的检验工作和产品质量控制提供科学依据和技术支持。
1 实验部分
1.1 仪器,试剂与材料
Thermo TSQ Vantage液相色谱-串联质谱仪(美国Thermo公司);乙腈、甲醇、甲酸、乙酸铵均为色谱纯(德国Merck公司);无水硫酸镁(分析纯,天津富宇试剂公司);N-丙基乙二胺吸附剂PSA(天津博纳艾杰尔公司);实验用水为实验室自制,符合GB/T 6682一级水的标准。
化学降糖药物标准品:泼尼松、氢化可的松、曲安西龙双醋酸酯、倍他米松、氟米龙、氟氢缩松、甲基泼尼松龙醋酸酯、氢化可的松丁酸酯、氢化可的松戊酸酯、倍他米松双丙酸酯购于中国食品药品检定研究院。
实验用保健品产品共计30份,均购于石家庄某超市。
1.2 标准储备液制备
分别将10种糖皮质类化合物用乙腈配制成质量浓度为1mg/mL的标准储备液,使用时用乙腈稀释成所需的标准工作溶液,-20℃冰箱保存。
1.3 样品处理
称取1.0g(精确至0.01g)样品于50mL具塞离心管中,加入乙腈10mL,涡旋混匀,再加入2.0g无水硫酸镁和100mgPSA,涡旋混匀后超声萃取20min,于4℃、10000r/min条件下离心5min,收集上清液并移至圆底烧瓶,42℃减压蒸发至近干。以10% 乙腈水溶解浓缩物,定容至1.0mL,过0.22μm 滤膜,供上机测定。
1.4 仪器条件
1.4.1 色谱条件:
色谱柱:Thermo Hypersil Gold(50mm×2.1mm,1.9μm)
流动相:A相0.1%(V/V)甲酸水溶液;B相乙腈;梯度洗脱:0-1min,90%A;1-8min,90%-10%A;8-10min,10%A;10-11min,10%A-90%A;11-14min,90%A。流速:0.30mL/min;柱温:30℃;进样量:1μL。
1.4.2 质谱条件:
电喷雾离子源(ESI)温度:350℃;
毛细管电压:4000V(正离子模式);
扫描范围:m/z 100~800(正离子模式);
气化温度:300℃;
鞘气压(N2):30arb;
辅助气压(N2):10arb;
离子传输管温度:350℃;
碰撞裂解气:高纯氩气;
2 结果与讨论
2.1 样品提取方法的优化
实验采用有机溶剂进行超声提取,比较了乙醇、甲醇、乙腈、三氯甲烷等4种有机溶剂,以确定本实验用的提取有机溶剂。分别选用以上四种溶剂作为提取剂和不同的提取时间(5、10、15、20、30min),对实际样品进行加标回收实验(加标量为10μg/kg),比较回收率、本底干扰及可操作性综合评价提取溶剂的效果。实验结果表明:乙腈的提取回收率最高,基质干扰最小,效果最好。当超声时间为20min时,目标物提取回收趋于稳定,均能获得70-115%的回收率。因此实验最终选择乙腈为提取溶剂,超声时间为20min。
2.2 净化条件的优化
由于保健品中用乙腈提取所得提取物基质较为复杂,基质效应明显,因此需对提取液进行净化处理,实验考察对比了固相萃取柱法和基质固相分散法的两种净化效果,以基质加标回收率的高低作为萃取净化方法评价指标。结果表明,使用HLB固相萃取柱净化得到了较好的效果,10种糖皮质激素平均回收率达到75%以上,降低了基质效应;使用基质固相分散萃取法,能够有效地去除样品基质中的色素、脂类、醚类、金属离子等杂质,回收率与使用HLB固相萃取相比,平均回收率无明显差异,但是平均回收率RSD值较高,这可能是由于PSA填料结构为N-丙基乙二胺,对一些糖皮质激素具有吸附作用,但是与使用固相萃取柱HLB相比,基质分散固相萃取操作更加简便,使用的提取剂用量少,操作更加简便,因此本实验最终选取PSA净化方法。
2.3 质谱条件的优化
根据10种糖皮质激素化合物的化学电离性质,分别选用正模式扫描和负模式扫描作为离子化模式,采用流动注射泵连续进样方式进行质谱条件的优化。结果表明,在实验结果表明10种糖皮质类激素物质在正模式和负模式均有响应,在正模式下形成[M+H]、[M+Na]峰、在负模式下形成[M-H]峰,其中10种糖皮质激素在正模式下的[M+H]峰的丰度最高,因此本实验确定以正模式扫描,以[M+H]来确定10种糖皮质激素的母离子,在确定糖皮质激素的母离子后,采用子离子扫描方式对子离子进行了优化选择,每个母离子确定了2对子离子。通过优化碰撞能量、透镜电压等质谱参数使每种激素的特征碎片离子产生的离子对强度达到最大。确定碰撞能量和透镜电压后,连接液相色谱和三重四级杆质谱仪联机,对离子源温度、毛细传输管温度、鞘气流量、辅助气流量进行优化,使样液中每种激素的离子化效率达到最佳。10种激素的质谱分析参数见表1。
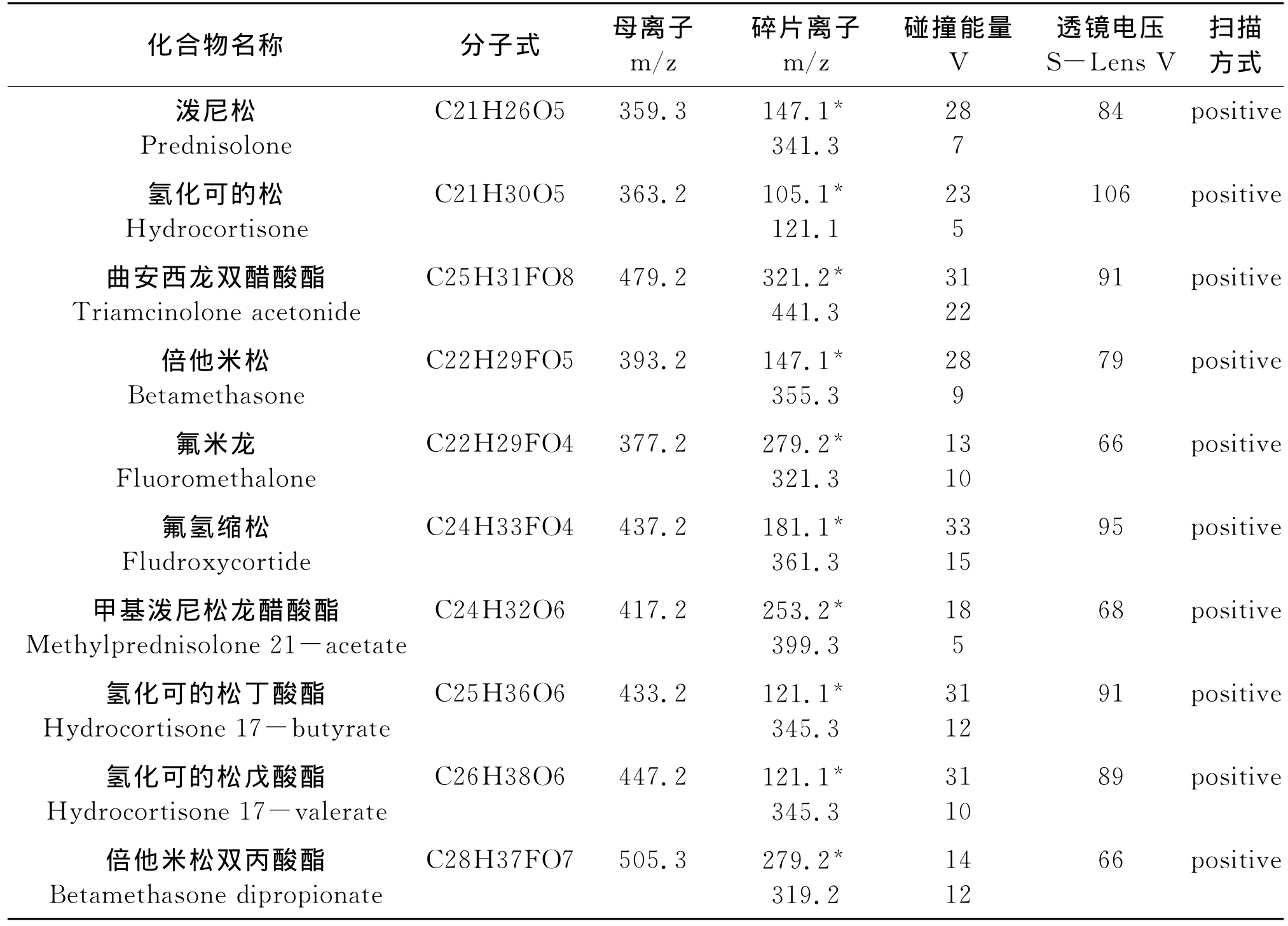
表1 10种糖皮质激素的质谱分析参数
2.4 色谱流动相的选择
液相色谱质谱仪在正模式下常用的流动相体系有甲酸水溶液(甲酸含量0.1%v/v)-甲醇体系、甲酸水溶液(甲酸含量0.1%v/v)-乙腈体系、乙酸铵水溶液(乙酸铵含量5mmol/L)-乙腈体系、乙酸铵水溶液(乙酸铵含量5mmol/L)-甲醇体系等,考察了以上四种流动相体系下,质谱出峰的情况。经过试验表明,不同流动相对质谱出峰影响较大,其中有机相使用乙腈体系得到的峰影响强度均大于甲醇体系,且基质干扰明显小于使用甲醇体系,水相中使用乙酸铵比使用甲酸得到的峰形更好,这可能是因为使用乙酸铵有利于目标物的分子离子化,改善化合物的峰形。但是使用乙酸铵水溶液作为水相得到的10种糖皮质激素平均峰强度比使用甲酸水溶液得到的10种糖皮质激素平均峰强度小1/2左右,这可能是由于甲酸使正离子模式下的目标物质子化增强,但同时也增加了糖皮质激素类物质在流动相中的保留能力,导致峰形变宽。综合考虑目标物的以上几点,本实验最终确定以甲酸水溶液(甲酸含量0.1%v/v)-乙腈体系作为流动相,优化后的目标物MRM色谱图见图1。
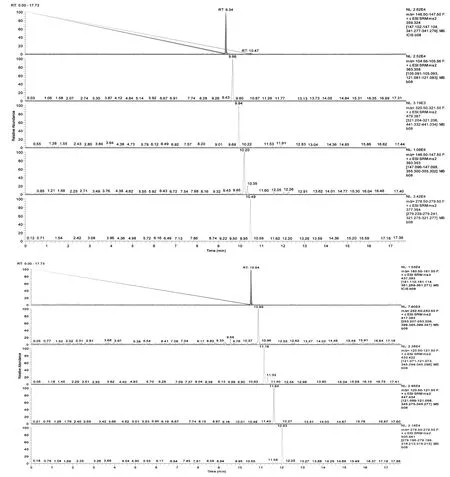
图1 10种糖皮质激素的MRM色谱图
(从上到下依次为泼尼松Prednisolone、氢化可的松Hydrocortisone、曲安西龙双醋酸酯Triamcinolone acetonide、倍他米松Betamethasone氟米龙Fluoromethalone、氟氢缩松Fludroxycortide、甲基泼尼松龙醋酸酯Methylprednisolone 21-acetate、氢化可的松丁酸酯Hydrocortisone 17-butyrate、氢化可的松戊酸酯Hydrocortisone 17-valerate、倍他米松双丙酸酯Betamethasone dipropionate)
2.5 方法准确度和精密度
以10种糖皮质激素物质标准品的峰面积为纵坐标,相应的质量浓度(ng/mL)为横坐标,进行线性回归计算,得到线性方程和相关系数。10种分析物的线性方程相关系数均大于0.99。以3倍信噪比(S/N)为检出限(LOD),10倍信噪比(S/N)为定量限(LOQ)。各目标物的线性范围、相关系数、检出限、定量限见表2。
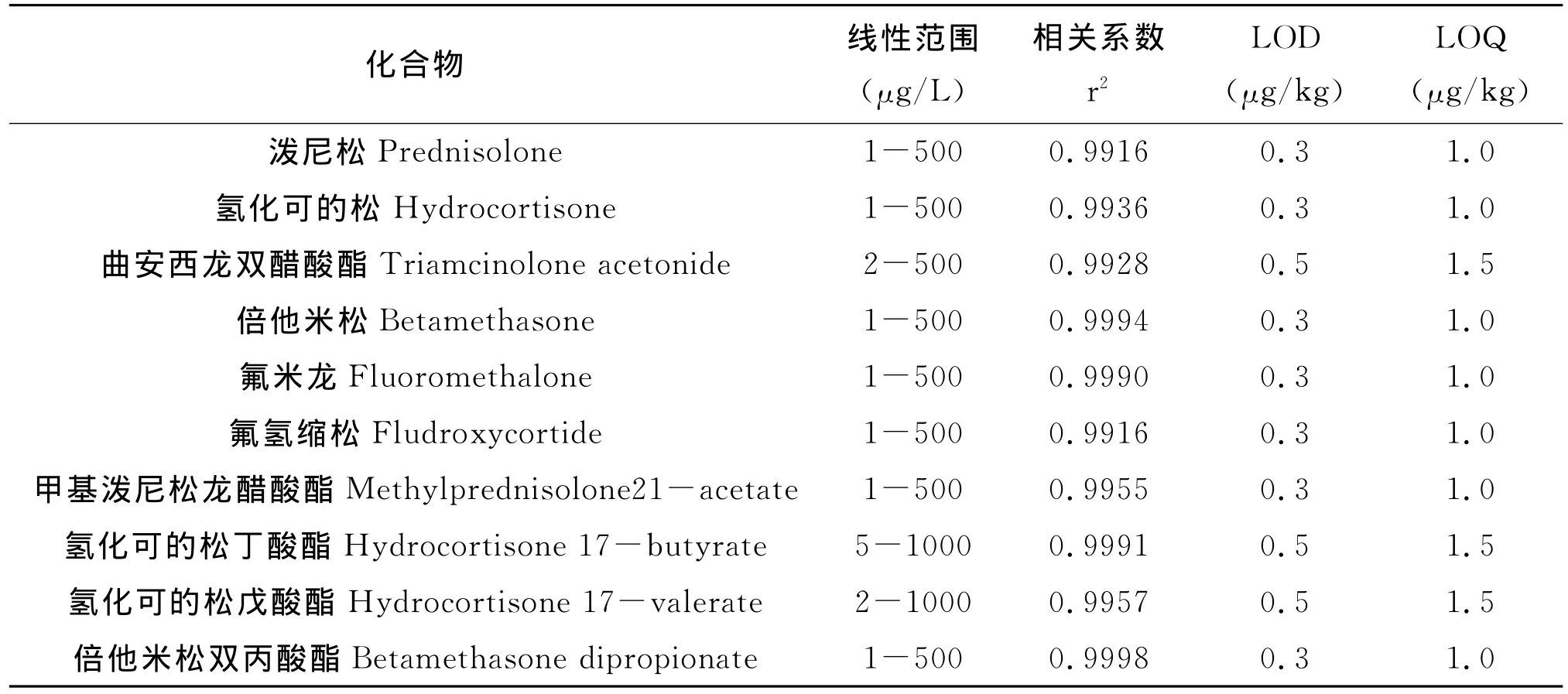
表2 10种目标物的线性范围、相关系数、检出限和定量限
2.6 方法的回收率
在空白基质中添加10种降压药物标准溶液,添加水平分别为1μg/kg、3μg/kg和10μg/kg,每个添加水平平行测定3次,用超高效液相色谱-串联质谱测定,其回收率和精密度结果列于表3。10种目标物在3种添加水平下的加标回收率在71.4%~113.5%之间,相对标准偏差为2.1%-13.6%。
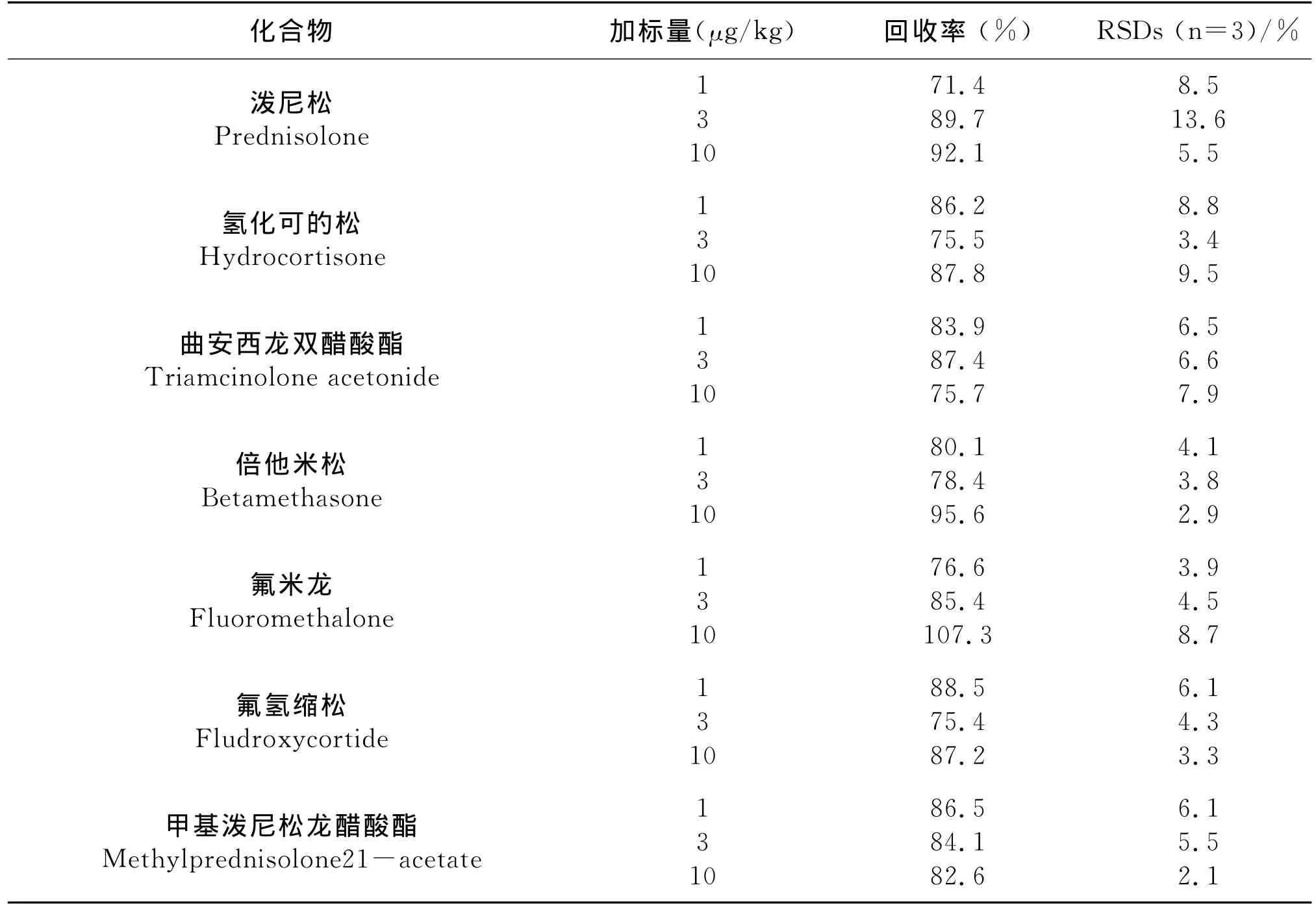
表3 实际样品的加标回收率和相对偏差
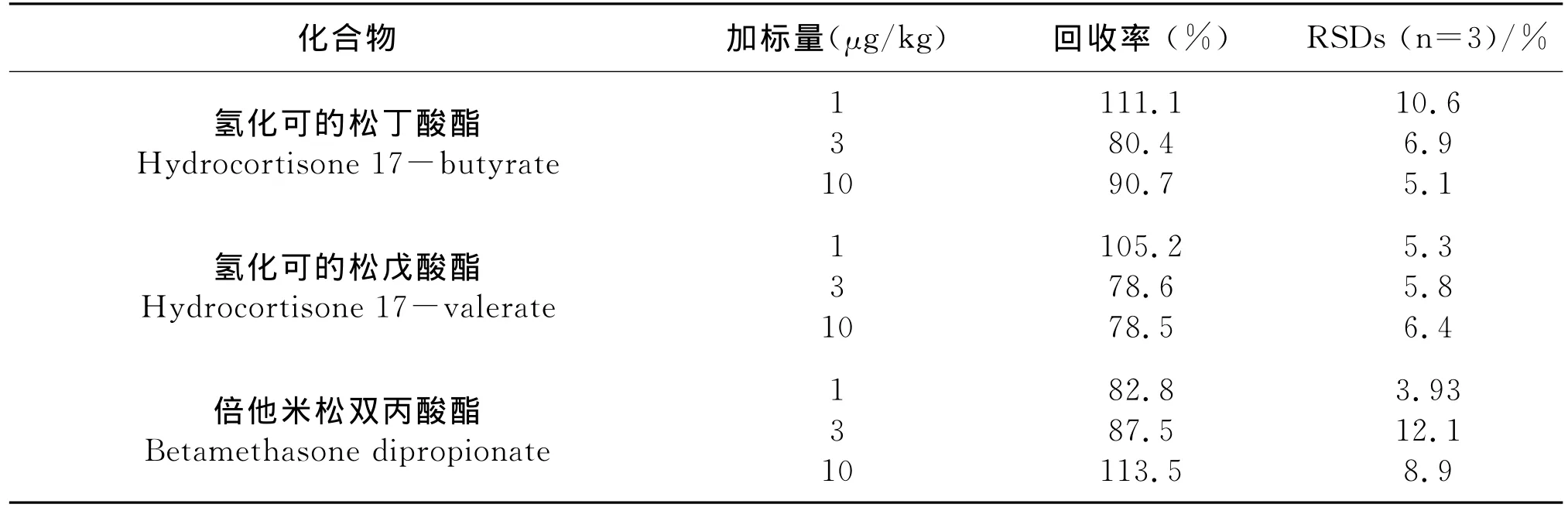
续表3
2.7 实际样品的测定
应用本方法对市场上销售的保健类产品共30份进行了测定。结果发现1份功能性保健食品中含有0.20mg/kg的倍他米松。
3 结论
建立了同时快速测定降糖类保健食品中的10种违禁添加成分的UPLC-MS/MS方法,通过优化液相条件和质谱条件,有效地提高了分析效率和灵敏度,缩短了检测的时间(在6min内能够完成10种药物的检测),降低了检出限,扩大了线性范围。方法的样品前处理步骤简单,有效降低了基质效应,回收率符合方法学要求。该方法可以用于保健食品中糖皮质激素类物质非法添加的检测。
[1] 杜昆.肾上腺皮质激素的不良反应[J].首都医药,2000,7(8):62-63.
[2] 李佩佩,郭远明,张小军,等.高效液相色谱-串联质谱法测定动物源食品中同化激素的研究进展[J].理化检验-化学分册,2014,50(11):1476-1477.
[3] 吴大南,郑和辉,王萍,等.超高效液相色谱法检测化妆品中8种糖皮质激素[J].中国卫生检验杂志,2008,18(2):197-198.
[4] 潘军.HPLC法筛查中药软膏制剂中非法添加的17种糖皮质激素[J].中成药,2014,36(11):2323-2324.
[5] 吴小红,李焕德.高效液相色谱法二极管阵列检测器同时分析测定中成药及保健品中非法添加的9种糖皮质激素[J].中南药学,2009,7(5):324-325.
[6] 孙雪婷,商少明,陈秀英,等.微乳毛细管电色谱电动进样-场放大堆积法检测化妆品中糖皮质激素[J].分析化学,2014,42(1):36-40.
[7] 夏瑞,董素英,车宝泉.薄层色谱法快速鉴别中药制剂中的糖皮质激素[J].药物分析杂志,2008,28(3):470-471.
[8] 吴维群,沈朝烨,杨玉林,等.GC-MS联用技术检测水性化妆品性激素成分的方法研究[J].环境与职业医学,2004,21(4):307-309.
[9] 王朝,马强,王星,等.液相色谱-串联质谱法同时测定化妆品中16种激素[J].分析化学,2007,35(9):1257-1262.
[10] 凌霄,徐志洲,牛冲.HPLC/MS/MS法检测中药制剂中解热镇痛及糖皮质激素类化学药[J].药物分析杂志,2010,30(7):1311-1314.
[11] 严华,云环,刘鑫,等.UHPLC-LTQ-OrbitrapMS测定鸡肉组织中5种糖皮质激素残留[J].分析测试学报,2013,32(8):909-914.
