阿归养血颗粒质量标准研究
2014-05-04龙海燕李文莉
赵 勇,王 艳,龙海燕,丁 野,李文莉
(1.湖南省食品药品检验研究院,湖南 长沙 410001; 湖南中医药大学药学院,湖南 长沙 410208)
阿归养血颗粒收载于《卫生部药品标准·中药成方制剂(第十二册)》[1],由当归、党参、白芍、甘草、茯苓、黄芪、熟地黄、川芎、阿胶九味药组方,具有补养气血的作用,用于气血亏虚、面色萎黄、眩晕乏力、肌肉消瘦、经闭、赤白带下等症候。原标准中仅收载了专属性不强的化学鉴别和当归的薄层色谱(TLC)鉴别。为进一步控制药品内在质量,保证临床用药安全、有效,笔者对其质量标准进行了深化研究,修订了当归的鉴别,增加了方中白芍、黄芪、甘草的TLC鉴别,并建立了采用高效液色谱(HPLC)法测定白芍中芍药苷含量的方法。现报道如下。
1 仪器与试药
LC-2010A型高效液相色谱仪(日本岛津公司);CG-300型超声波清洗器(300 W,25 kHz,江苏张家港港威超声波仪器厂),AE-240型电子天平(梅特勒-托利多仪器上海有限公司)。阿魏酸对照品(批号为110773-200611)、芍药苷对照品(批号为110736-200629)、甘草对照药材(批号为 120904-200511)、甘草苷对照品(批号为111610-200604)、黄芪甲苷对照品(批号为110781-200613),均购自中国药品生物制品检定所;阿归养血颗粒由国内4家生产企业提供,共8批样品,批号分别为061102,061103, 061104, 060901, 06125001, 20060306, 20060307,20060308;乙腈为色谱纯,水为纯化水,其他试剂均为分析纯。
2 方法与结果
2.1 HPLC法鉴别[2]
2.1.1色谱条件
色谱柱:Hypersil BDS C18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈-0.1%磷酸溶液(13∶87);检测波长:323 nm;流速:1.0 mL/min;柱温:40℃。
2.1.2溶液制备
取含量测定项下的供试品溶液,作为供试品溶液;取阿魏酸对照品适量,加甲醇制成每1 mL含20 μg的溶液,即得对照品溶液;按处方称取缺当归和川芎的其他药材,按制备工艺制成缺当归和川芎的阴性样品,同供试品溶液制备方法制成双阴性对照品溶液。
2.1.3测定法
分别精密吸取对照品溶液5 μL、供试品溶液和双阴性对照品溶液各15 μL,注入高效液相色谱仪,依法测定,记录色谱图。供试品溶液色谱中,出现与对照品溶液主峰保留时间一致的色谱峰,缺当归和川芎的双阴性对照品溶液无干扰(见图1)。
2.2 TLC鉴别
白芍[3]:取本品50 g,研细,加甲醇70 mL,超声处理30 min,放冷,滤过,滤液蒸干,残渣加水30 mL使溶解,用以水饱和的正丁醇振摇提取2次,每次30 mL,合并正丁醇液,用氨试液洗涤2次,每次30 mL,弃去碱液,取正丁醇液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取芍药苷对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。再按处方比例称取除白芍外其他药材适量,制成缺白芍的阴性样品,同法制成缺白芍的阴性对照品溶液。照TLC法[2010年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各15 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(45∶5∶10∶0.2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,缺白芍的阴性对照无干扰。见图2A。
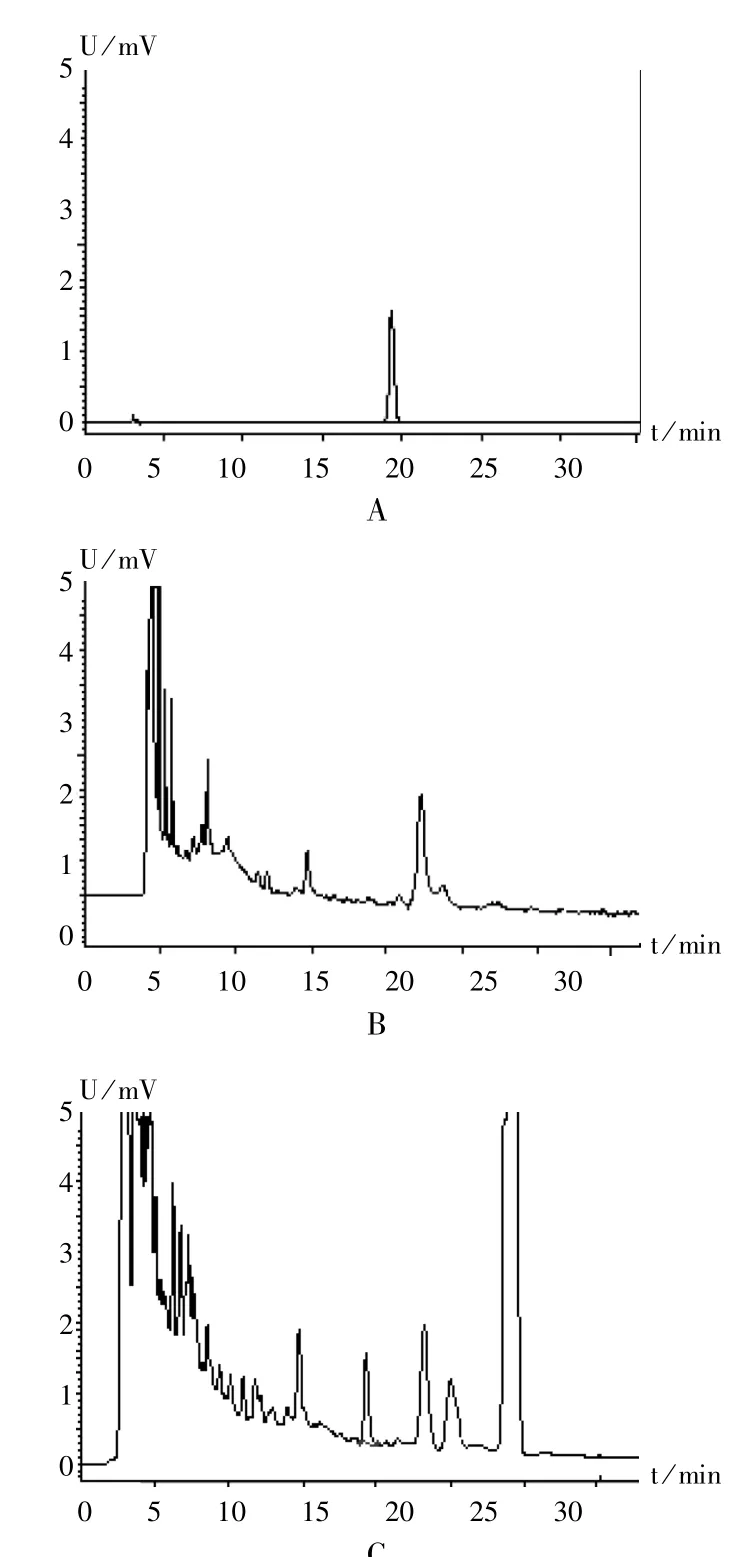
图1 当归和川芎高效液相色谱鉴别图
甘草[4]:取本品25 g,研细,加甲醇50 mL,超声处理30 min,放冷,滤过,滤液蒸干,残渣加水30 mL使溶解,用以水饱和的正丁醇振摇提取2次,每次30 mL,合并正丁醇液,用以正丁醇饱和的水洗涤2次,每次30 mL,取正丁醇液蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。另取甘草对照药材0.5 g,加甲醇25 mL,超声处理20 min,放冷,滤过,滤液蒸干,残渣加甲醇2 mL使溶解,作为对照药材溶液。再取甘草苷对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。另按处方比例称取除甘草外其他药材适量,制成缺甘草的阴性样品,同法制成缺甘草的阴性对照品溶液。照TLC法[2005年版《中国药典(一部)》附录VI B]试验,吸取上述4种溶液各5~10 μL,分别点于同一含1%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干,喷以 10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照药材和对照品溶液色谱相应位置上显相同颜色的斑点,缺甘草的阴性对照无干扰。见图2B。
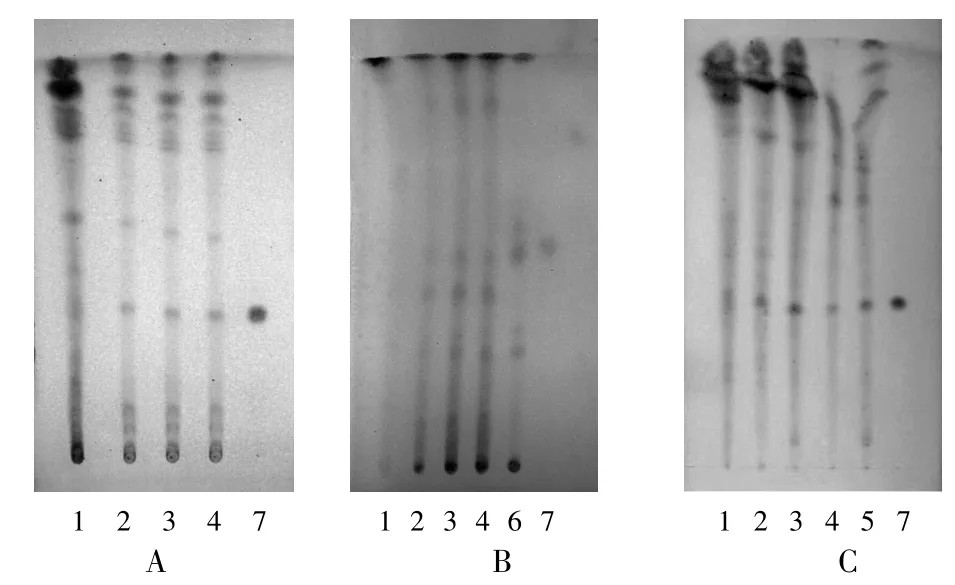
图2 薄层色谱图
黄芪[5]:取黄芪甲苷对照品适量,加甲醇制成每1mL含0.5 mg的溶液,作为对照品溶液。另按处方比例称取除黄芪外其他药材适量,制成缺黄芪的阴性样品,同甘草鉴别项下供试品溶液制备方法制成缺黄芪的阴性对照品溶液。照TLC法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取对照品溶液、甘草鉴别项下供试品溶液及缺甘草的阴性对照品溶液各15 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)10℃以下放置12 h的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,缺黄芪的阴性对照无干扰。见图2C。
2.3 芍药苷含量测定[6-9]
2.3.1色谱条件
色谱柱:Agilent TC-C18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈-水(10∶90);柱温:35 ℃;流速:1.0 mL/min;检测波长:230 nm。
2.3.2溶液制备
取芍药苷对照品适量,精密称定,加甲醇制成40 μg/mL的溶液,即得对照品溶液。取装量差异项下的本品适量,研细,取约5 g,精密称定,置锥形瓶中,精密加甲醇25 mL,称定质量,密塞,超声处理15 min(功率300 W,频率25 kHz),放冷,称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。取缺白芍的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。
2.3.3方法学考察
系统适用性试验:分别吸取2.3.2项下3种溶液各10 μL,注入高效液相色谱仪,记录色谱图,见图3。供试品溶液色谱中芍药苷能达到基线分离,分离度符合要求,理论板数按芍药苷峰计算应不低于5 000,阴性对照无干扰。
线性关系考察:精密吸取芍药苷对照品溶液(48.56 μg/mL)1,2,5,10,15,20 μL,注入高效液相色谱仪,依法测定,以峰面积积分值为纵坐标(Y)、进样量(X,μg)为横坐标绘制标准曲线,得回归方程 Y=1 194 685.842X+9 458.073 505,r=0.999 9(n=6)。结果表明,芍药苷对照品进样量在0.048 56~0.971 2 μg范围内与峰面积呈良好线性关系。
精密度试验:精密吸取同一对照品溶液10 μL,连续进样6次。结果芍药苷峰面积的 RSD为0.5%(n=6),表明仪器精密度良好。
重复性试验:取同一批(批号为110736-200629)样品,按拟订方法制备6份供试品溶液,依法测定。结果平均含量为0.208 7 mg/g,RSD为1.1%(n=6),表明方法重复性良好。
稳定性试验:取同一供试品溶液,分别于配制后的0,3,6,9,12,15 h时依法测定。结果芍药苷峰面积的 RSD为1.0%(n=6),表明供试品溶液在15 h内稳定。

图3 芍药苷含量测定高效液相色谱图
加样回收试验:取已测定含量的同一批(批号为20060306)样品(含芍药苷0.208 7 mg/g)约2.5 g,精密称定,置锥形瓶中,精密加入对照品溶液(0.485 6 mg/mL)1 mL,置水浴上蒸干,放冷,精密加入甲醇25 mL,依法制备供试品溶液,测定并计算。结果见表1。
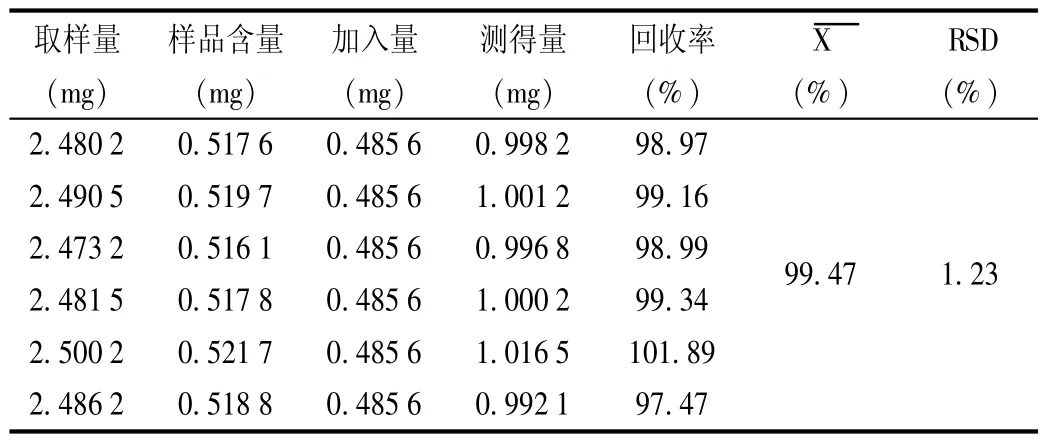
表1 芍药苷加样回收试验结果(n=6)
2.3.4样品含量测定
按拟订方法测定8批样品的含量。结果见表2。
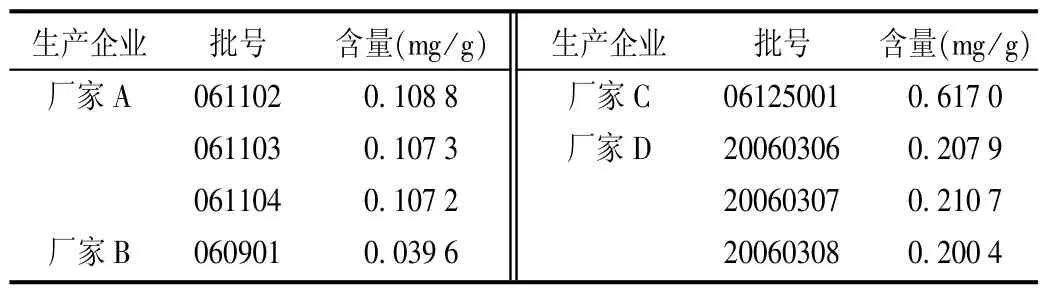
表2 样品含量测定结果
3 讨论
原标准中收载了当归的TLC鉴别,经试验验证发现,缺当归的阴性对照有干扰,经分析干扰主要来自方中的川芎,两者在现有TLC条件下很难区分,故采用HPLC法鉴别两者中的阿魏酸并作为质量控制指标。
白芍的TLC鉴别曾考察了将展开剂中毒性较大的三氯甲烷改为毒性较小的二氯甲烷,结果所得供试品TLC图效果较差,斑点较模糊。
甘草的TLC鉴别时比较了乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)、三氯甲烷-甲醇-水(30∶10∶1)2种展开剂。结果以前者分离效果较好,斑点清晰,能得到甘草中主要的4个黄色主斑点,阴性对照无干扰。
黄芪的TLC鉴别时,曾考察了供试品溶液的制备方法,先用甲醇回流提取,后用水饱和的正丁醇振摇提取,最后过中性氧化铝柱,结果所得TLC图斑点较模糊,分析原因可能为黄芪甲苷在过中性氧化铝柱时有部分损失。另外,也考察了将展开剂中毒性较大的三氯甲烷改为毒性较小的二氯甲烷,结果所得供试品TLC图效果较差,Rf值较高。
含量测定项曾考虑以阿魏酸作为测定指标,但根据各药味的处方用量及2010年版《中国药典(一部)》[10]各药材项下含量测定规定限度,按理论值推算的阿魏酸含量低于芍药苷,且该成分稳定性不如芍药苷,故选择白芍中芍药苷为定量指标成分。
芍药苷的含量测定时,比较了甲醇、80%的甲醇、50%的甲醇3种提取溶剂,以及加热回流和超声处理2种提取方法,结果所测得芍药苷的含量基本一致,但以甲醇超声处理方法操作简单,所得供试品溶液黏度小、颜色浅、易于滤过,故选择该方法作为供试品溶液的制备方法。流动相比较了甲醇-0.05%冰醋酸(25∶75)、乙腈-0.1% 磷酸溶液(13∶87)、乙腈-水(13∶87)3 种流动相,结果以流动相乙腈-水(13∶87)所得色谱峰对称性较好,且对色谱柱的损伤较小,故选择乙腈-水(13∶87)为流动相。
4个生产厂家提供的样品中芍药苷的含量相差极大,分析原因可能为原法定标准中无芍药苷的定量项目,某些企业存在偷工减料、少投或以次充好等违法现象。为了更好地控制产品的质量,保证临床用药安全有效,有必要建立芍药苷的含量测定项。
参考文献:
[1]WS3-B-2338-97,卫生部药品标准·中药成方制剂(第十二册)[S].
[2]辛继跃,邓强华.HPLC法测定当归南枣片中阿魏酸的含量[J].江西中医药,2011,42(8):61-62.
[3]胡颖菲,陆兔林,毛春芹,等.大柴胡颗粒质量标准研究[J].中成药,2013,35(1):68-73.
[4]杨志福,曹 军,杨 静,等.咽炎咀嚼片质量标准的建立[J].中国药师,2012,15(3):327-329.
[5]陈海云,吴 王君,袁 易,等.肠胃清口服液的薄层色谱鉴别[J].中国药业,2012,21(1):28-29.
[6]杜天信,李 洁.HPLC法测定桃仁膝康丸中芍药苷的含量[J].中国医药指南,2012,10(3):10-11.
[7]郑美善,徐莲花,崔永学.高效液相色谱法测定止痛清晕胶囊中芍药苷的含量[J].中国药业,2012,21(1):34.
[8]陈 黎,彭红霞,刘春霞,等.HPLC法测定固肾调经片中芍药苷的含量[J].中国药师,2012,15(12):1 741-1 743.
[9]李捷玮,金柔男,李 翔,等.多种中成药中芍药苷的含量测定[J].药物分析杂志,2009,29(9):1 440-1 446.
[10]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:38,96,124.
