抗磷脂综合征57例临床分析
2014-04-20许菡苡牛晓婷
许菡苡,赵 伟,胡 红,牛晓婷,白 雪
解放军总医院,北京 100853 1呼吸科;2风湿免疫科
抗磷脂综合征57例临床分析
许菡苡1,赵 伟2,胡 红1,牛晓婷1,白 雪1
解放军总医院,北京 1008531呼吸科;2风湿免疫科
目的分析抗磷脂综合征(antiphospholipid syndrome,APS)的临床表现、诊断及治疗,以提高本病的诊治水平。方法回顾性分析2004 - 2013年在本院诊断明确的57例APS患者的临床特征和免疫学特点。结果57例中男性14例(24.6%),女性43例(75.4%),确诊时平均年龄(37±14)岁;原发性APS 21例(36.8%),继发性APS 36例(63.2%)。血栓发生率71.9%,血栓复发率41.5%,原发性APS的血栓复发率高于继发性APS(P<0.05);病态妊娠发生率63.3%。本组患者65.4%抗心磷脂抗体(anticardiolipin antibodies,ACL)阳性,71.9%抗β2-糖蛋白Ⅰ抗体阳性,42%血小板减少。在确诊APS前,43.9%被诊断为血栓事件,33.3%被诊断为自身免疫性疾病,8.8%被诊断为血小板减少。结论APS临床表现以血栓形成、病态妊娠为主,免疫学特点为高滴度的ACL和(或)抗β2-糖蛋白Ⅰ抗体阳性。APS临床症状多样化导致其误诊率较高。
抗磷脂综合征;血栓形成;抗β2-糖蛋白Ⅰ抗体
抗磷脂综合征(antiphospholipid syndrome,APS)是以血栓形成、病态妊娠为主要临床表现,伴有抗心磷脂抗体(anticardiolipid antibody,ACL)、抗β2-糖蛋白Ⅰ(anti-β2glycoproteinⅠantibodies,β2-GPⅠ)抗体或狼疮抗凝物(lupua anticoagulants,LA)阳性为特征的自身免疫性疾病[1]。APS的主要病理基础是血栓形成,APS发病机制尚不清楚,可能与以下因素有关:1)抗磷脂抗体介导的内皮细胞激活及血管内皮损伤;2)影响抗凝及纤溶系统导致细胞聚集和血栓形成;3)部分可能与机体遗传基因易感有关[2]。APS可分为原发性和继发性,原发性APS病因尚不清楚,可能与遗传、感染等因素有关,而继发性APS常伴随其他疾病,如系统性红斑狼疮等[1,3]。此外,还有一种少见的灾难性抗磷脂综合征,表现为短期内进行性广泛血栓形成,造成多器官功能衰竭甚至死亡[4]。由于APS较为少见,临床表现多样化,病情反复,容易漏诊误诊。本研究拟通过对在本院确诊且资料完整的57例APS患者的临床表现、实验室检查、诊断及治疗进行分析,以提高对本病的认识及诊治水平。
资料和方法
1 资料 2004年1月- 2013年8月本院住院确诊的57例APS患者,均符合2006年Sydney国际诊断标准[1]。
2 方法 收集患者年龄、性别、伴发疾病、自初始症状到确诊的时间、临床表现、ACL、抗β2-糖蛋白Ⅰ抗体、狼疮抗凝物、血小板、治疗情况及随访资料等。
3 统计学方法 应用SPSS13.0统计软件,计量资料结果以±s差表示,采用t检验;计数资料采用χ2检验,P<0.05为差异有统计学意义。
结 果
1 患者一般情况 57例中,男性14例(24.6%),女性43例(75.4%),男女比例约为1∶3。确诊时年龄14 ~ 70(37±14)岁。从初始症状到确诊APS的中位时间为30个月,其中>60个月16例(28.1%)。原发性APS共21例(36.8%),继发性APS共36例(63.2%);继发性APS中97.2%继发于自身免疫性疾病(35/36)。
2 血栓事件 APS血栓发生率71.9%(41/57),以下肢深静脉血栓、脑梗死、肺栓塞为多见(表1)。血栓事件中静脉血栓51.2%(21/41)、动脉血栓19.5%(8/41)、静脉血栓合并动脉血栓29.3%(12/41)。53.7%(22/41)为单一部位血栓形成,29.2%(12/41)两个部位血栓形成,17.1%(7/41) 3个以上部位血栓形成。58.5%患者出现单次血栓,41.5%(17/41)患者出现反复血栓(≥2次),其中70.6%(12/17)血栓复发的血管类型与初次受累时相同。原发性APS的血栓复发率高于继发性APS(P<0.05)。APS合并血小板减少患者的血栓发生率低于不合并血小板减少者(OR=0.21,P=0.006 5)。
3 病态妊娠 30例女性有婚育史,发生病态妊娠19例(63.3%)。其中,自发性流产8例(42.1%),胎儿发育异常5例(26.3%),宫内死胎2例(10.5%),早产2例(10.5%),重度子痫前期2例(10.5%)。
4 受累系统 1)免疫系统:自身免疫性疾病35例,其中系统性红斑狼疮30例、干燥综合征1例、类风湿关节炎1例、系统性硬化1例、皮肌炎1例、未分化结缔组织病1例。2)血液系统:血小板减少24例,贫血5例。3)肾表现:狼疮肾炎12例,肾病综合征3例,慢性肾功能不全3例,左肾动脉栓塞1例,肾小球肾炎1例。4)皮肤表现:皮肤紫癜及网状青斑6例,雷诺现象4例,肢体溃疡2例,干性坏疽1例。5)呼吸系统:肺栓塞5例,其中2例反复发生肺栓塞(≥2次)者出现了肺动脉高压;5例间质性肺炎。6)神经系统:脑梗死8例,颅内静脉窦血栓形成2例。7)心脏表现:心脏瓣膜病变4例,心肌梗死1例。8)消化系统:腹腔静脉血栓形成2例,门静脉血栓形成1例。9)骨:缺血性骨坏死2例,趾骨坏死1例。10)眼:左眼视网膜中央动脉阻塞2例。见表2。
5 实验室检查 ACL阳性率为65.4%(34/52,余5例未查),抗β2-糖蛋白Ⅰ抗体阳性率为71.9% (41/57),ACL和抗β2-糖蛋白Ⅰ抗体同时阳性率为48.1%(25/52,余5例未查ACL)。7例行狼疮抗凝物检测,其中5例阳性。42%(24/57)患者血小板减少,其中29.2%(7/24)<50×109/L。
6 误诊情况 在确诊APS前,25例(43.9%)仅诊断为血栓事件,其中肢体血栓事件17例、脑梗死5例、肺栓塞3例。19例(33.3%)仅诊断为自身免疫性疾病,5例仅诊断为血小板减少(8.8%),3例仅诊断为贫血(5.3%)。
7 治疗及预后 抗血小板药物阿司匹林治疗21例;肝素治疗25例,其中低分子肝素19例、普通肝素6例;这些患者均过渡到华法林钠抗凝治疗。溶栓治疗6例。糖皮质激素治疗48例,环磷酰胺、甲氨蝶呤、羟氯喹等免疫抑制剂治疗42例。输血治疗12例,输血小板11例。静脉丙种球蛋白治疗16例。心脏瓣膜置换术2例,颅内横窦球囊扩张术1例,下肢静脉滤器置入术治疗4例。57例经治疗后均好转出院。随访至今,30例规律用药后控制良好;15例血栓复发患者中7例住院治疗,3例死亡(2例因系统性红斑狼疮、1例因重症肝损伤);12例失访。
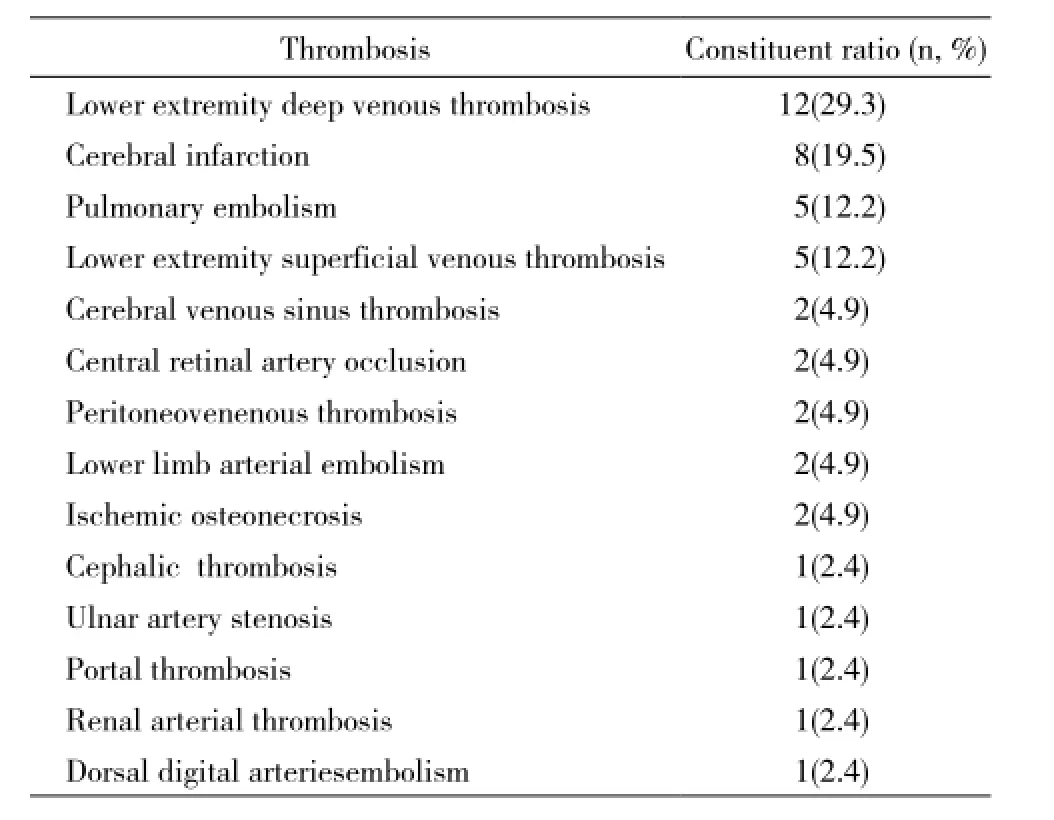
表1 APS患者的血栓事件Tab. 1 Occurrence of thrombosis in APS patients (n=41)
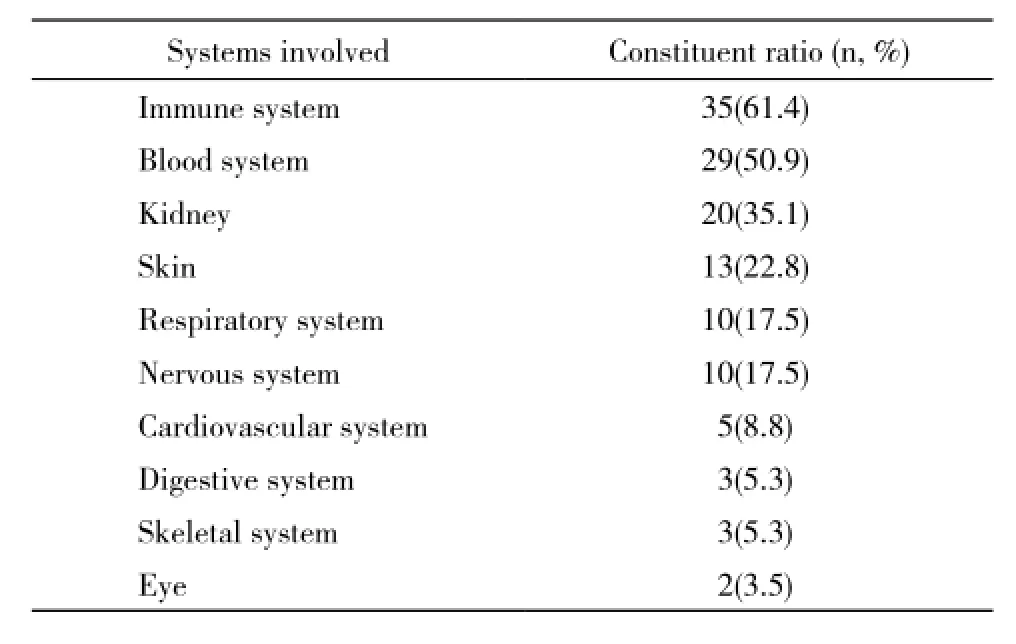
表2 APS患者受累系统和器官Tab. 2 Involved systems and organs in APS patients (n=57)
讨 论
本组APS患者的临床特征为血栓发生率71.9%,病态妊娠发生率63.3%,71.9%患者抗β2-糖蛋白Ⅰ(β2-GPⅠ)抗体阳性,65.4%患者抗心磷脂抗体阳性。最新修订的APS诊断标准为必须同时具备至少1项临床标准和1项实验室标准[1]。
APS血栓形成有以下特点:1)血栓可发生于全身各部位,所有血管均可累及。本组病例中,71.9%患者发生血栓事件,其中静脉血栓51.2%、动脉血栓19.5%,静脉血栓合并动脉血栓29.3%。Cervera等[4]报道来自13个欧洲国家的1 000例APS患者中37.1%出现静脉血栓,27.0%出现动脉血栓,15.2%静脉血栓合并动脉血栓,与该数据比较,本组患者静脉血栓发生率较高而动脉血栓较低,可能与种族差异或样本量较少有关。2)血栓可在单一部位发生,也可在多部位发生。3)血栓容易反复发作。本组中58.5%患者出现单次血栓,而41.5%患者出现2次或以上的血栓事件,其中70.6%(12/17)血栓复发的血管部位及类型与初次受累时相同。Muñoz-Rodriguez等[5]报道在100例APS患者中47.2%出现单次血栓事件,52.8%有反复血栓栓塞,86%在同一部位血栓形成复发,其反复血栓发生率高于本组,可能与其原发性APS比例(62%)较高有关,本组结果显示原发性APS的血栓复发率明显高于继发性APS。
病态妊娠是APS另一项突出的临床特征,主要表现为习惯性流产、胎儿发育异常、宫内死胎、早产及重度子痫前期等。有文献报道在590例女性APS患者中35.4%流产,16.9%胎死宫内,略高于本组数据[4]。病态妊娠可能是由于ACL、抗β2-糖蛋白Ⅰ抗体诱导胎盘血栓形成、干扰种植导致胎盘发育不良、胎盘梗死甚至流产及胎死宫内[3-6]。因此,建议习惯性流产妇女进行抗磷脂抗体检测,以利于APS早期诊断。
APS除上述典型临床表现外,还可因血栓事件累及不同的血管导致全身多个系统器官受累。本组资料显示APS可累及免疫系统、血液系统、肾、皮肤、呼吸系统、神经系统、心血管系统等进而出现复杂多样的临床表现。Cervera等[4]报道APS使全身多个系统器官受累出现相应的临床症状有可能出现误诊、漏诊。在确诊APS前,本组43.9%患者仅被诊断为血栓事件,33.3%患者仅被为诊断为自身免疫性疾病,8.8%仅被诊断为血小板减少。本组从初始症状到确诊APS所需的中位时间为30个月,其中>60个月的患者16例(28.1%)。Donnan和Mcdonald[7]在伦敦所进行的一项调查显示,从初始症状到确诊APS所需的中位时间为30个月,甚至有患者的误诊时间长达50年。上述结果提示,因为APS症状多样化、临床表现复杂,受累器官较多,致使部分患者确诊时间较长,误诊、漏诊较多,因此须提高内科、全科及专科医生对APS的认识。
抗β2-糖蛋白Ⅰ抗体、ACL、LA均为APS的独立实验室指标。本组71.9%患者抗β2-糖蛋白Ⅰ阳性,高于国内外的文献报道[8-9]。抗β2-糖蛋白Ⅰ抗体是2006年Sydney诊断标准中新增指标,是抗磷脂抗体的主要靶抗原,该抗体诊断APS的特异性高于ACL[1]。本组ACL和抗β2-糖蛋白Ⅰ抗体同时阳性率为48.1%,文献报道联合检测抗磷脂抗体可提高APS的检出率[9]。
血小板减少亦为APS常见的临床表现之一,但缺乏特异性,自身免疫性疾病、感染、药物等均可导致血小板减少。血小板减少机制可能为:ACL直接作用于血小板膜蛋白,破坏血小板,导致血小板减少;磷脂-蛋白-抗磷脂抗体三分子复合物介导的血栓形成及血小板减少[10]。本研究中24例(42.1%)患者出现血小板减少。本组单因素分析显示,合并血小板减少的APS患者血栓发生率明显低于不合并血小板减少者,考虑血小板减少可能是防止血栓形成的有利因素。
APS目前尚无标准化治疗方案。治疗原则主要为防治血栓、控制出血、治疗并发症、对症处理和改善预后[11-13]。有高危因素而无症状患者可使用抗血小板聚集药物阿司匹林75~81 mg/d。静脉血栓患者抗凝治疗原则:初始使用普通肝素或低分子量肝素至少5 d,与华法林钠治疗重叠后改用口服华法林钠,并使国际标准化比值(INR)达到2.0 ~ 3.0。对于抗凝治疗时间,应当依据个体化栓塞事件严重程度、是否存在高凝因素以及潜在的出血并发症等综合考虑,推荐长期甚至终生抗凝治疗。糖皮质激素与免疫抑制剂不作为常规治疗,但在出现灾难性APS时,除常规抗凝治疗外,强调应用糖皮质激素、静脉应用丙球蛋白、血浆置换以及环磷酰胺等免疫抑制疗法[14-15]。此外,自身免疫性疾病导致的继发性APS应针对原发病给予糖皮质激素和(或)免疫抑制剂治疗。对血小板<50×109/ L的APS患者可考虑应用泼尼松1 ~ 2 mg/(kg·d),联合大剂量丙球蛋白治疗,但应禁用抗凝治疗。
综上所述,APS以血栓事件、病态妊娠为主要临床表现,可累及全身多个系统器官,以高滴度的ACL和(或)抗β2-糖蛋白Ⅰ抗体和(或)LA阳性为免疫学特征。临床上常因对APS症状的复杂性、多样性认识不足而导致误诊或漏诊。如果发现血栓事件、病态妊娠的患者,应及时检查ACL、抗β2-糖蛋白Ⅰ抗体或LA,使APS得以早期诊治。
1 Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS)[J]. J Thromb Haemost, 2006,4(2): 295-306.
2 Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome[J]. N Engl J Med, 2013, 368(11):1033-1044.
3 中华医学会风湿病学分会. 原发性抗磷脂综合征诊治指南(草案)[J]. 中华风湿病学杂志, 2003, 7(9): 574-576.
4 Cervera R, Piette JC, Font J, et al. Antiphospholipid syndrome:clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients[J]. Arthritis Rheum,2002, 46(4): 1019-1027.
5 Muñoz-Rodriguez FJ, Font J, Cervera R, et al. Clinical study and follow-up of 100 patients with the antiphospholipid syndrome[J]. Semin Arthritis Rheum, 1999, 29(3): 182-190.
6 Ruiz-Irastorza G, Crowther M, Branch W, et al. Antiphospholipid syndrome[J]. Lancet, 2010, 376(9751):1498-1509.
7 Donnan PT, Mcdonald MJ. Patients’ experiences of a diagnosis of Hughes’ syndrome[J]. Clin Rheumatol, 2009, 28(9): 1091-1100.
8 陈晓微,申艳,孙传银,等.抗磷脂综合征174例临床特点及与欧洲数据的比较分析[J].中华风湿病学杂志,2010,14(6):394-397.
9 Gardiner C, Hills J, Machin SJ, et al. Diagnosis of antiphospholipid syndrome in routine clinical practice[J]. Lupus, 2013, 22(1):18-25.
10 Giannakopoulos B, Passam F, Rahgozar S, et al. Current concepts on the pathogenesis of the antiphospholipid syndrome[J]. Blood,2007, 109(2): 422-430.
11 Lim W. Antiphospholipid antibody syndrome[J]. Hematology Am Soc Hematol Educ Program, 2009:233-239.
12 Giannakopoulos B, Krilis SA. How I treat the antiphospholipid syndrome[J]. Blood, 2009, 114(10): 2020-2030.
13 George D, Erkan D. Antiphospholipid syndrome[J]. Prog Cardiovasc Dis, 2009, 52(2):115-125.
14 Sciascia S, Lopez-Pedrera C, Roccatello D, et al. Catastrophic antiphospholipid syndrome (CAPS)[J]. Best Pract Res Clin Rheumatol, 2012, 26(4): 535-541.
15 Aguiar CL, Erkan D. Catastrophic antiphospholipid syndrome:how to diagnose a rare but highly fatal disease[J]. Ther Adv Musculoskelet Dis, 2013, 5(6): 305-314.
Antiphospholipid syndrome: A clinical analysis of 57 patients
XU Han-yi1, ZHAO Wei2, HU Hong1, NIU Xiao-ting1, BAI Xue1
1Department of Respiratory Medicine;2Department of Rheumatology Chinese PLA General Hospital, Beijing 100853, China
Corresponding author: HU Hong. Email: huhong_dr@aliyun.com
ObjectiveTo improve the diagnosis and treatment of antiphospholipid syndrome (APS) by analyzing its clinical manifestations, diagnosis and treatment.MethodsClinical and immunological features of 57 APS patients admitted to our hospital from 2004 to 2013 were retrospectively analyzed.ResultsOf the 57 APS patients, 14 (24.6%) were males and 43 (75.4%) were females with an average age of (37±14) years, 21 (36.8%) were diagnosed with primary APS and 36 (63.2%) were diagnosed with secondary APS. The incidence of thrombosis was 71.9% and its recurrence rate was 41.5%. The recurrence rate of thrombosis was higher in primary APS patients than in secondary APS patients (P<0.05). The incidence of abnormal pregnancy was 63.3%. The anticardiolipin antibodies (ACL) and anti-β2glycoproteinⅠantibodies were positive in 65.4% and 71.9% APS patients, respectively. The incidence of thrombocytopenia was 42%. Before APS was diagnosed, 43.9%, 33.3% and 8.8% patients were misdiagnosed as thrombosis, autoimmune diseases and thrombocytopenia, respectively.ConclusionAPS is mainly manifested as thrombosis and abnormal pregnancy with positive ACL and/or anti-β2glycoproteinⅠantibodies, and is thus frequently misdiagnosed.
antiphospholipid syndrome; thrombosis; anti-β2glycoproteinⅠantibodies
R 593.2
A
2095-5227(2014)06-0541-04
10.3969/j.issn.2095-5227.2014.06.006
2014-03-07 14:47
http://www.cnki.net/kcms/detail/11.3275.R.20140307.1447.007.html
2014-01-07
中华医学会临床医学科研专项资金(08020350113)
许菡苡,女,在读硕士。研究方向:哮喘与COPD。Email: yixu360@126.com
胡红,女,主任医师,教授,博士生导师。Email: huho ng_dr@aliyun.com
