RNA病毒感染对应激颗粒的调控作用
2014-04-13王晓旭孙英杰丁云磊王桂军
王晓旭,孙英杰,丁云磊,王桂军,丁 铲
(1.安徽农业大学动物科技学院,合肥 223006;2. 中国农业科学院上海兽医研究所,上海 200241;3. 南京农业大学动物医学院,南京 210095)
·综述·
RNA病毒感染对应激颗粒的调控作用
王晓旭1,孙英杰2,丁云磊3,王桂军1,丁 铲2
(1.安徽农业大学动物科技学院,合肥 223006;2. 中国农业科学院上海兽医研究所,上海 200241;3. 南京农业大学动物医学院,南京 210095)
哺乳动物细胞受到热休克、氧化应激、营养缺乏或者病毒感染等环境压力时,能够迅速启动细胞的压力应答机制,终止细胞内的蛋白翻译,在这个过程中,往往会形成应激颗粒。应激颗粒作为胞浆中终止活动的翻译起始复合物的聚集产物,在细胞的基因表达和内平衡中发挥着重要的作用。尤其是当病毒感染细胞时,应激颗粒的形成可以使细胞的蛋白翻译水平大大降低,从而抑制病毒的复制。然而在病毒的长期进化过程中,也衍生出了对抗细胞压力应答的相应机制,有些病毒甚至可以利用应激颗粒中包裹的沉默的转录本促进自身的复制。本文将着重就RNA病毒对应激颗粒的调控以及最近提出的压力应激与先天性免疫之间的关系做一综述。
应激颗粒;RNA病毒;调控机制;先天性免疫
当哺乳动物细胞遇到环境压力时可以迅速形成一种可逆的动态结构—应激颗粒(stress granules, SGs),它能够降低细胞整体的翻译速率。SGs的形成是从停滞组装的43S和48S核糖体起始复合物的聚集开始的,它作为一个临时储存库来存放这些复合物,信使核糖核蛋白(messenger ribonucleo protein, mRNP)向应激颗粒中的聚集增加了细胞的存活几率,而当压力解除时,这些翻译复合物可以迅速被释放并恢复蛋白质的合成,从而恢复细胞内稳态。
1 应激颗粒的形成机制
最常见的触发应激颗粒形成的条件有氧化压力、营养缺乏或热应激压力,此时一种eIF2α激酶被激活,这些激酶包括:血红素调节抑制激酶(heme-regulated inhibitor kinase,HRI)、总蛋白调节激酶(general control non-derepressible kinase,GCN2)、双链RNA依赖性激酶(protein kinase R,PKR)或PKR样内质网激酶(PKR-like endoplasmic reticulum kinase,PEKR),促使真核翻译起始因子2(eukaryotic initiation factor 2, eIF2)的α亚基被磷酸化,阻止了eIF2B将eIF2-GDP转变成eIF2-GTP,从而将eIF2B滞留于eIF2复合物之中,进一步减少了翻译起始复合物eIF2-GTP-Met-tRNAMet[1,2],抑制细胞的翻译过程,造成大量核糖核蛋白的结合聚集,最终形成应激颗粒。其中,不同的蛋白激酶分别被不同的压力源激活:当细胞处于热应激或缺铁情况时,HRI被激活;当细胞营养缺乏时,蛋白激酶GCN2被活化;当细胞面对蛋白平展或折叠错误的压力时,PEKR则被激活;而通常病毒感染会因其双链RNA的识别而触发PKR的活化,从而引起eIF2α的磷酸化[3]。除了依赖于eIF2α的磷酸化介导应激颗粒的形成之外,也有的是通过抑制真核翻译起始因子4G(eukaryotic initiation factor 4G, eIF4G)或者真核翻译起始因子4A(eukaryotic initiation factor 4A, eIF4A)的功能来抑制蛋白的翻译,从而诱导应激颗粒的聚集[4-7]。
应激颗粒形成的分子机制包括以下几个步骤:几种关键组分RNA结合蛋白的自身低聚化;蛋白翻译后的修饰;mRNP在微管上的运输。应激颗粒包涵了数百种RNA互作的蛋白,更有研究显示超过一百种基因参与了SGs的装配,因此SGs形成的机制是多种多样且相当复杂的[8],但仅仅携带了有限的基因的简单的病毒却进化出了能够有效控制应激颗粒形成与功能的机制。
2 SGs成分的多样性
应激颗粒的形成是从停滞的翻译起始复合物的聚集开始的,典型的应激颗粒以几种关键的翻译起始因子(例如eukaryotic initiation factor 4E,eIF4E等)、mRNA和40S核糖体亚基的高浓度聚集为形成标志[9-11]。此外,还有许多RNA结合蛋白(如Y box binding protein 1,YB1等),可能只要是能够与RNA结合的蛋白,或者可以强烈的与mRNP互作的蛋白都会存在于SGs中。这些蛋白大多数都只是暂时储存于SGs中,并无特殊的生物学功能与意义。应激颗粒中也包含了一些与它的形成密切相关的重要的标志蛋白,特别是G3BP1(Ras GTPasactivating protein-binding protein 1)、TIA1(T-cell intracellular antigen 1)、TIAR(TIA-related protein)、TDRD3(tudor domain containing 3)、HDAC6(histone deacetylase 6)和Caprin1[12-16]。迄今为止的研究结果表明:在研究病毒与应激颗粒的相互作用过程中,一些标志蛋白在细胞质中的聚集,并不一定就是应激颗粒,即某一两种应激颗粒的标志蛋白的聚集,并不能断定为具有功能的应激颗粒的形成[17]。
在应激颗粒中有许多标志性成分,这些成分与其形成以及功能具有很大的联系,并且存在于各种压力刺激下产生的颗粒中,但是应激颗粒的其他组成成分却会因为不同的压力刺激而不同。例如,热休克引起的应激颗粒(HS-SGs)中特异性存在热休克蛋白27(heat shock protein 27,hsp27),而这个蛋白在亚砷酸钠诱导的SGs(Ars-SGs)中就不存在[9,13,18]。亚硒酸盐诱导的SGs中存在大多数典型的翻译因子,但却唯独缺乏翻译起始因子eIF3b[19]。病毒感染引起细胞压力十分特殊,在这类病毒感染性应激颗粒(V-SGs)中存在Sam68蛋白,而此蛋白在HS-SGs中并没有发现[18]。由此可见,尽管应激颗粒作为细胞压力应答机制的重要组成部分,存在于各类哺乳动物细胞中,发挥着相似的功能,但其组成成份却不尽相同。
3 病毒对应激颗粒的调节机制
病毒感染细胞后干扰了宿主的多项进程,从而激活了细胞的压力应答。尽管许多病毒在早期感染确实可以引起应激颗粒,但是大多数病毒则可以在某些感染周期中抑制应激颗粒的产生。另外值得一提的是,在已知的例子中,几乎不存在具有功能的应激颗粒(包涵有停滞翻译的复合物)的细胞内,病毒仍然可以高效率的复制。这意味着病毒和应激颗粒之间存在着一种敌对性的关系,这也更加便于我们理解人们对应激颗粒的功能性定义—具有翻译沉默和RNA降解的功能的蛋白聚集颗粒。
前面讨论到应激颗粒的形成方式多种多样,因此不难推出病毒对应激颗粒的调节也存在着许多不同的方式。为了便于理解,下面的讨论将根据病毒与应激颗粒互作的机制不同分成以下三类:病毒对PKR-eIF2α通路的调控;病毒对SGs核心蛋白的“挟持”;病毒对SGs组分的切割[20]。如图1所示,病毒对SGs的调控主要通过抑制PKR的磷酸化、劫持或切割TIA/TIAR、G3BP等SGs核心蛋白来抑制SGs的形成。而其中有一些病毒,虽然可以促进eIF2α的磷酸化,但是也可以通过一些其他未知机制来抑制SGs的形成。由于这些互作大部分是对病毒系统的初步探索,有些甚至存在着争议,因此这种分类还有待验证。
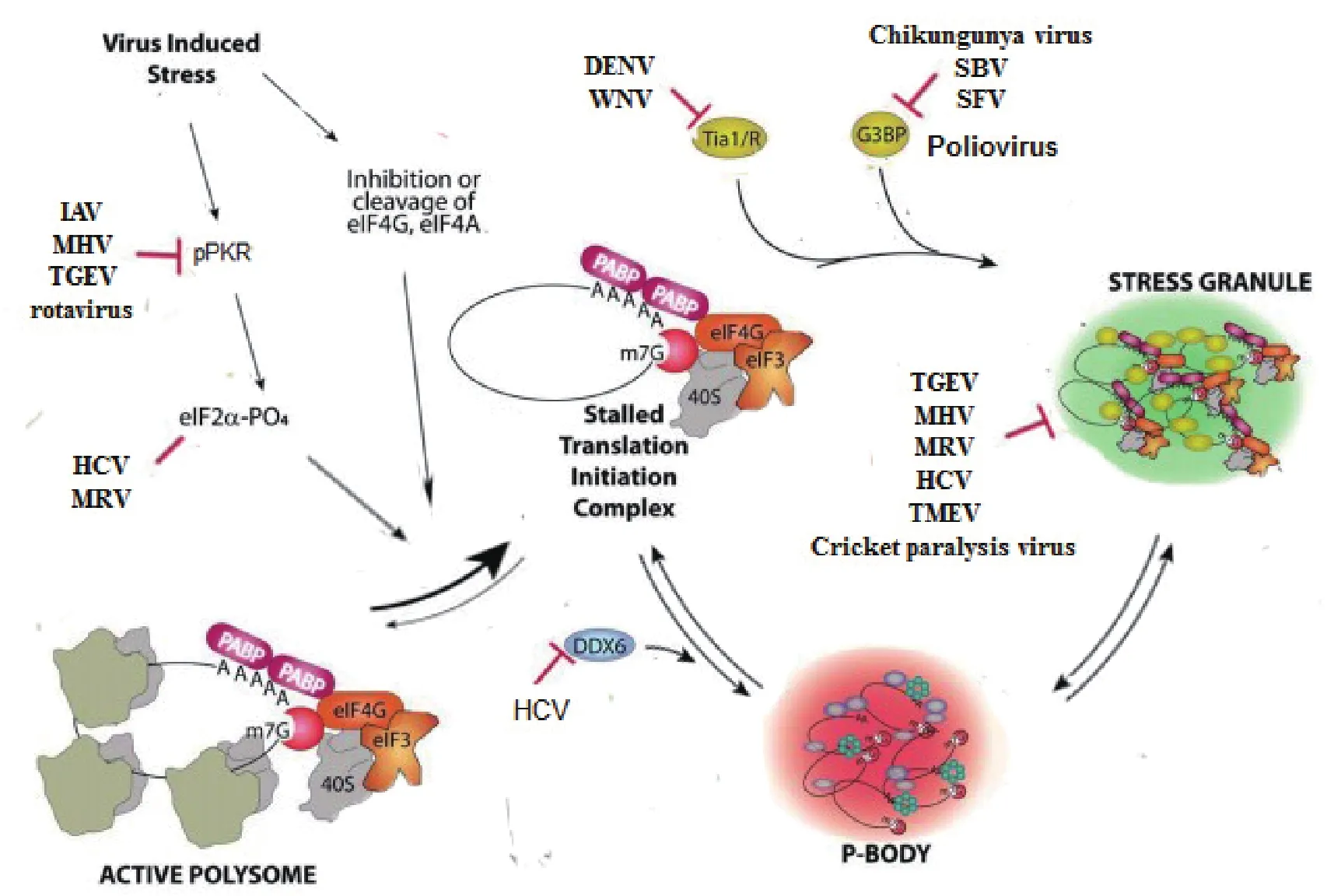
图1 病毒对SGs的调控作用[21]Fig. 1 The modulation of virus to SGs
3.1 病毒对PKR-eIF2α通路的调控PKR是细胞在受到压力刺激和病毒感染时激活压力应答和先天性免疫的一个重要的传感器。大多数的动物病毒都可以触发PKR的活化,并且许多病毒也存在着抵抗PKR活性的机制,这些机制也会对应激颗粒的形成产生影响。PKR的激活和由此产生的翻译抑制可以强烈诱导SGs形成,但是SGs通过G3BP诱导的mRNP的聚集也引起PKR的活化[7]。这可能是一些病毒也可通过折叠蛋白反应来激活PERK,并同样导致下游SGs形成,但这之间的直接联系目前尚未见报道。
在普通感染情况下,A型流感病毒(Influenza A virus,IAV)可以通过病毒非结构蛋白NS1的活化来阻止应激颗粒的产生,而NS1蛋白可被自身具有双链RNA绑定结构域活化,从而抑制PKR磷酸化的作用。然而,对不能绑定双链RNA的NS1蛋白突变体的表达,却可以引起eIF2α的磷酸化,并引起SGs的聚集。当SGs形成后,通过对病毒蛋白NP的检测,可以得知,病毒的复制受到了抑制。此外,用NS1突变的病毒感染PKR敲除的细胞系并不能形成应激颗粒[22],这表明了NS1对PKR的活性的抑制,是IAV抑制应激颗粒形成的关键。同样,用缺失NS1的病毒感染细胞可以形成应激颗粒。NS1蛋白对应激颗粒的抑制机制包括了它与细胞一个包括RNA结合蛋白55(RNA-associated protein 55,RAP55)的复合体的互作[23]。RAP55既存在于应激颗粒中也存在于P小体中,并且可能促进mRNP在他们之间的穿梭功能。过表达RAP55可以引起应激颗粒,并且抑制病毒的复制。当NS1蛋白缺失时,病毒的NP蛋白与应激颗粒共定位,但是在野生型病毒感染时,则转而与P小体共定位。一些有争议的报道显示:使用缺失了NS1的突变体病毒感染细胞,仍然引起了PKR的活化,eIF2α的磷酸化和应激颗粒的形成,但是NP蛋白的复制并没有发生改变[24]。这表明了可能不仅仅是不同种的应激颗粒存在组成成份的差异,即使是同种病毒感染不同种类型的细胞也会存在差异(A549细胞与HeLa细胞)。
轮状病毒的病毒蛋白P2、nsP2和nsP5可以积极促进PKR和eIF2α的磷酸化,从而控制宿主细胞的翻译系统,但这种磷酸化并不影响病毒自身蛋白的合成[25]。用亚砷酸钠刺激感染了轮状病毒的细胞并不能观察到应激颗粒的形成,由此可见,轮状病毒的感染可以抵制细胞受到的外源性压力刺激,但这之间的作用机制尚不明确。
哺乳动物正呼肠孤病毒(Mammalian orthoreovirus, MRV)在感染早期(6 h)可以诱导应激颗粒的形成,使得eIF2α的磷酸化增强,并使细胞内整体的蛋白翻译受到抑制[26]。有研究表明,MRV诱导的SGs的形成并非依靠某一种蛋白激酶的活化来促进eIF2α的磷酸化,而是通过多条途径激活该反应[27]。而在MRV感染的晚期,虽然仍旧能使eIF2α的磷酸化,但是SGs的数量明显减少,并且细胞开始优先翻译病毒的mRNA[26,27]。很有可能MRV存在着另外一条不依赖eIF2α的磷酸化的通路,但这种猜测还有待验证。
丙型肝炎病毒(Hepatitis C virus,HCV)同样是依赖eIF2α的磷酸化诱导应激颗粒的形成[28,29]。HCV复制极其缓慢,并且可以持续改变其对应激颗粒的调控方式:一边组装,一边拆卸,使得细胞内的蛋白表达处于抑制和未抑制的动态变化之中。对于应激颗粒的拆卸,HCV是通过GADD34介导的eIF2α的去磷酸化来实现的[28]。
传染性胃肠炎病毒(Transmissible gastroenteritis virus,TGEV)和小鼠肝炎冠状病毒(Mouse hepatitis virus,MHV)均属于冠状病毒属,在早期感染过程中均可形成包含有TIAR的应激颗粒[30,31]。目前还没有证据表明TGEV可以在感染后期分解应激颗粒。在干扰了SGs的组分PTB后,病毒的蛋白的复制增加,证明应激颗粒的产生可以抑制TGEV的复制[31]。同样,在PKR S51A突变体小鼠成纤维细胞中,MHV的复制也显著增强[30]。
3.2 病毒对SGs核心蛋白的“挟持”大多数的应激反应都可以从多个层次来调节细胞的基因表达,其中最显著的是调控蛋白翻译和RNA降解。在最初期的病毒蛋白合成后,正链RNA病毒必须将个体基因组从一个招募核糖体的状态转变成抑制翻译的状态,以此将核糖体从模板上清除,以便于RNA的复制。因此,在病毒复制过程中,细胞质中存在着多种RNA调控蛋白也就不足为奇了。如果说应激颗粒的一些关键因子,如G3BP1、TIA1必须聚集在一起,才能促使应激颗粒形成,那么,病毒就可以通过对这些宿主因子的调控,来重新改变宿主利用这些因子所想达到的功能和效果。
一些甲病毒属病毒的非结构蛋白可以与G3BP1互作,形成复合物[32,35],例如塞姆里斯森林病毒(Semliki forest virus,SFV)。在SFV感染早期可以使eIF2α磷酸化,诱导应激颗粒的形成[36],而到了后期则可以抑制其形成。SFV的nsP3蛋白可以将G3BP1蛋白包裹在病毒复制复合物中,从而抑制了应激颗粒的形成[37]。与之相似,Chikungunya病毒的nsP3蛋白也同样可以通过招募G3BP1蛋白到新型的细胞质颗粒中来抑制应激颗粒的形成。这两种病毒nsP3蛋白与G3BP1与的互作结构域均存在于其羧基端,但其具体位置却不同。另外,SFV在靠近翻译起始密码子的区域存在翻译增强子,可以逃避eIF2α磷酸化导致的翻译抑制作用,并且有利于拆分应激颗粒[35,37]。
辛德毕斯病毒(Sindbis virus,SBV/SINV)同样属于甲病毒属,其RNA依赖的RNA聚合酶nsP4蛋白可以与G3BP1和G3BP2互作[32],而G3BP1又可以与nsP2和nsP3互作,因此就形成了一个复杂的病毒蛋白酶复合物[33,34]。在缺乏G3BP1的情况下,病毒的RNA水平并没有发生较大的改变,但病毒多聚蛋白水平却显著上升。这表明了G3BP1对病毒翻译的影响远远大于对RNA复制的影响。
黄病毒属的西尼罗河病毒(West Nile virus,WNV)和登革热病毒(Dengue virus,DENV)同样可以抑制亚砷酸钠诱导的SGs的形成[38]。而这种“挟持”机制涉及了多个SGs成核的关键蛋白,如TIA1、TIAR和G3BP1。研究表明,西尼罗河病毒不能诱导SGs产生的原因很有可能是赋予了TIAR新的功能——促进自身病毒复制[39,40]。病毒的3'端的茎环结构可以与TIA1和TIAR结合,并与细胞核周围的复制酶富集区共定位,以此来促进病毒的复制[38]。由此可见,病毒对SGs成核的关键蛋白的“挟持”可以防止应激颗粒的形成,促进自身的复制。
之前提到的HCV可以通过调控eIF2α的磷酸化来控制应激颗粒,同样,它也可以通过对应激颗粒成分的“挟持”来调控应激颗粒的形成。HCV可以在细胞质形成新型的蛋白聚集,这其中包括了G3BP1、DDX6、RCK/p54、Xrn1等[41],它们在感染后期与丙肝病毒的核心蛋白共定位成环状。而通过干扰DDX6、G3BP1等,HCV的复制水平也显著下降[17],这说明了病毒利用了应激颗粒及P小体的成分蛋白来帮助自身的复制。
3.3 病毒对SGs组分的切割许多的正链RNA病毒可以通过表达一些病毒蛋白酶来裂解宿主的一些重要的蛋白,以此来调节细胞内环境。迄今为止,只有肠病毒(如脊髓炎灰质病毒)已被证实能够利用蛋白酶来降解细胞RNA颗粒的相关因子。脊髓炎灰质病毒可以在感染的早期阶段形成病毒感染性应激颗粒(V-SGs),但到了病毒感染的中后期,病毒就会抑制并分解应激颗粒。应激颗粒的分解机制包括了病毒3C蛋白酶对应激颗粒成核蛋白G3BP1的切割。G3BP1的切割将其氨基端的蛋白互作结构域和羧基端的RNA识别区域分开,从而破坏了它对应激颗粒形成的聚集作用。在病毒感染后期,通过表达抑制3C蛋白剪切G3BP1的蛋白突变体可以产生应激颗粒,这表明了G3BP1在应激颗粒形成中的重要性[42]。而一些与之相矛盾的研究结果显示,在一些病毒刺激性应激颗粒中,直到病毒感染的后期仍然可以检测到应激颗粒的标志分子TIA1[18],而随后,人们发现在这些颗粒中往往缺乏翻译因子eIF3、eIF4G、eIF4E以及mRNA。这表明了在G3BP1被切割后残余的TIA1颗粒并不是传统意义上的富集了失速的翻译起始复合物,并能够抑制细胞翻译的应激颗粒[11]。因此,脊髓炎灰质病毒可能是通过切割G3BP1,来将TIA1从凝集了翻译起始因子的应激颗粒中拆分开来。这些发现说明了病毒感染引起的应激颗粒在功能和结构上都存在着各种差异,因此并不能仅仅通过对几个应激颗粒的标志分子的研究就得出可靠的结论。脊髓灰质炎病毒感染可以诱导eIF2α的磷酸化,与此同时可以通过诱导应激颗粒的形成来限制细胞的蛋白合成系统,从而抑制病毒RNA的复制。而有趣的是,脊髓炎灰质病毒可以通过3C蛋白酶切割eIF5B来越过在翻译起始时对eIF2α的依赖,但是这对于应激颗粒的形成并没有多大的影响[43]。
在小核糖核酸病毒总科中,有另外两种病毒也可以表达3C蛋白酶(具有不同的切割特异性),它们同样可以抑制应激颗粒的形成,但是并未有报道称是切割应激颗粒的重要蛋白来发挥这样的作用。Theiler氏鼠脑脊髓炎病毒(Theiler’s murine encephalomyelitis virus,TMEV)也可以通过一种未知的病毒蛋白来抑制应激颗粒的形成,并且在感染过程中这种蛋白能够保持G3BP1的完整性[44]。Cricket paralysis virus是小核糖核酸病毒亚族的成员之一,它也可以在不切割G3BP1和TIA-1的旁系同源基因(Rin-8 and Rox)的情况下,在感染后2 h就抑制应激颗粒的形成。病毒的3C蛋白酶在细胞受到压力的情况下是游离的,而在病毒感染情况下则并非如此,表明还有其他的病毒蛋白影响着它的亚细胞定位[45],也预示着可能还存在着其他未知的重要的应激颗粒蛋白分子被切割。
4 应激颗粒与先天性免疫
最近,有观点认为细胞面对病毒感染产生的压力环境时的压力应答在很多层面上都与先天性免疫具有着重要的联系。压力应答的过程中,或多或少都会有先天性免疫相关蛋白成分的参与,而这其中,有些蛋白成分还起到至关重要的作用。
PKR是一个典型的干扰素应答蛋白分子,它能够与病毒的RNA结合,感知细胞内病毒核酸的存在情况,介导下游的干扰素反应。与此同时,PKR的活化诱导了eIF2α的磷酸化,阻滞了细胞内的翻译进程,引起应激颗粒的产生。在对许多病毒的研究中,人们都发现缺失了PKR的MEF细胞失去了产生应激颗粒的能力,由此可以看出PKR在细胞压力应答中的重要地位。更有研究表明,PKR还参与细胞的生长调控,它可以磷酸化胰岛素受体基质1(insulin receptor substrate 1,IRS1)[46],从而参与细胞新陈代谢。此外,PKR在细菌和双链RNA病毒感染时,还可以激活炎性小体和巨噬细胞的应答作用,引起细胞因子IL-1b和HMGB1的释放[47]。在先天性免疫的转录应答中,PKR也发挥着重要作用[48-50]。由此可见,多种细胞内的营养应答与病原免疫反应都可以通过PKR这样的效应蛋白相互关联。
最新的研究表明,细胞的压力应答,往往会通过一些蛋白的聚集而与先天性免疫相关联。RIG-I样受体(RIG-I、MDA5、LGP2)是先天性免疫中识别RNA病毒的一类重要的模式识别受体,可以介导干扰素的产生,从而发挥抗病毒的作用。而在对细胞压力应答的研究中,人们发现在用亚砷酸钠刺激细胞后,RIG-I样受体可以进入应激颗粒之中。同样,用缺失了NS1的突变体流感病毒感染细胞后,RIG-I样受体同样可以被包裹入应激颗粒之中[24]。但这种互作和共定位能够发挥什么样的功能,目前仍旧不是十分清楚。在对缺失NS1的突变体流感病毒的研究中,甚至发现病毒的RNA也进入了应激颗粒之中[24]。而其他的病毒,如脊髓炎灰质病毒的RNA则不能进入病毒感染诱导的应激颗粒之中,在这之中,某些被突变的病毒蛋白(如流感NS1蛋白)可能对应激颗粒的形成、组分及功能发挥着重要的影响。
综合这类研究,我们不难发现,细胞的压力应答,在各种方面都与先天性免疫有着千丝万缕的联系,而这其中的机制目前仍旧不甚明朗,有待于后期的研究来揭示细胞中各类先天性免疫信号通路与细胞压力应答之间的联系及其在功能上的相互影响。
5 结语
在病毒感染引起的细胞各类应答反应中,压力应答仍旧是一类相对较新的领域。虽然目前已经有大量的关于各类病毒对应激颗粒操纵的报道,但是目前人们对这其中的作用机制仍然知之甚少。
当细胞面对压力环境时,细胞内的应答反应是广泛且复杂的,它能够动员起细胞内的各种成分来调控mRNA的活化与沉默,使之处于一种最优化的动态平衡之中。而病毒,作为引起细胞压力环境的强大的诱因,往往也能够迅速调集这些细胞内的变化情况,进化出多种机制抵抗细胞对它的抑制,甚至直接利用细胞的这种应激来帮助自身的复制。由于应激颗粒形成机制的复杂性,我们至今仍未能完全了解应激颗粒形成的具体过程。目前我们知道的关于病毒对应激颗粒的作用机制仅仅是由各类研究结果拼凑而成,并不能形成一个完整的机制系统。
此外,SGs形成后产生的各类压力信号,介导先天性免疫的抗病毒反应的机制也需要引起更多的重视。目前已知SGs在许多层面上都与先天性免疫相互关联,因此,在对于应激颗粒的研究中可能会发现一些在抗病毒治疗中具有价值的广谱的作用位点。
[1] Kedersha N, Chen S, Gilks N, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents ofmammalian stress granules[J]. Mol Biol Cell, 2002, 13(1): 195-210.
[2] Wek R C, Jiang H Y, Anthony T G. Coping with stress: eIF2 kinases and translational control[J]. Biochem Soc Trans, 2006, 34(Pt 1): 7-11.
[3] Montero H, Trujillo-Alonso V. Stress granules in the viral replication cycle[J]. Viruses, 2011, 3(11): 2328-2338.
[4] Dang Y, Kedersha N, Low W K, et al. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A[J]. J Biol Chem, 2006, 281(43): 32870-32878.
[5] Mazroui R, Sukarieh R, Bordeleau M E, et al. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation[J]. Mol Biol Cell, 2006, 17(10): 4212-4219.
[6] Emara M M, Fujimura K, Sciaranghella D, et al. Hydrogen peroxide induces stress granule formation independent of eIF2alpha phosphorylation[J]. Biochem Biophys Res Commun, 2012, 423(4): 763-769.
[7] Reineke L C, Dougherty J D, Pierre P, et al. Large G3BP-induced granules trigger eIF2alpha phosphorylation[J]. Mol Biol Cell, 2012, 23(18): 3499-3510.
[8] Ohn T, Kedersha N, Hickman T, et al. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly[J]. Nat Cell Biol, 2008, 10(10): 1224-1231.
[9] Kedersha N L, Gupta M, Li W, et al. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules[J]. J Cell Biol, 1999, 147(7): 1431-1442.
[10] Kedersha N, Stoecklin G, Ayodele M, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling[J]. J Cell Biol, 2005, 169(6): 871-884.
[11] White J P, Lloyd R E. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation[J]. J Virol, 2011, 85(23): 12442-12454.
[12] Tourriere H, Chebli K, Zekri L, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules[J]. J Cell Biol, 2003, 160(6): 823-831.
[13] Gilks N, Kedersha N, Ayodele M, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1[J]. Mol Biol Cell, 2004, 15(12): 5383-5398.
[14] Goulet I, Boisvenue S, Mokas S, et al. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules[J]. Hum Mol Genet, 2008, 17(19): 3055-3074.
[15] Kwon S, Zhang Y, Matthias P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response[J]. Genes Dev, 2007, 21(24): 3381-3394.
[16] Solomon S, Xu Y, Wang B, et al. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs[J]. Mol Cell Biol, 2007, 27(6): 2324-2342.
[17] Ariumi Y, Kuroki M, Kushima Y, et al. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets[J]. J Virol, 2011, 85(14): 6882-6892.
[18] Piotrowska J, Hansen S J, Park N, et al. Stable formation of compositionally unique stress granules in virusinfected cells[J]. J Virol, 2010, 84(7): 3654-3665.
[19] Fujimura K, Sasaki A T, Anderson P. Selenite targets eIF4E-binding protein-1 to inhibit translation initiation and induce the assembly of non-canonical stress granules[J]. Nucleic Acids Res, 2012, 40(16): 8099-8110.
[20] Lloyd R E. Regulation of stress granules and P-bodies during RNA virus infection[J]. Wiley Interdiscip Rev RNA, 2013, 4(3): 317-331.
[21] Lloyd R E. How do viruses interact with stress-associated RNA granules?[J]. PLoS Pathog, 2012, 8(6): e1002741.
[22] Khaperskyy D A, Hatchette T F, Mccormick C. Influenza A virus inhibits cytoplasmic stress granule formation[J]. FASEB J, 2012, 26(4): 1629-1639.
[23] Mok B W, Song W, Wang P, et al. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection[J]. J Virol, 2012, 86(23): 12695-12707.
[24] Onomoto K, Jogi M, Yoo J S, et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity[J]. PLoS One, 2012, 7(8): e43031.
[25] Montero H, Rojas M, Arias C F, et al. Rotavirus infection induces the phosphorylation of eIF2alpha but preventsthe formation of stress granules[J]. J Virol, 2008, 82(3): 1496-1504.
[26] Qin Q, Carroll K, Hastings C, et al. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2alpha phosphorylation and PKR[J]. J Virol, 2011, 85(17): 8798-8810.
[27] Qin Q, Hastings C, Miller C L. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection[J]. J Virol, 2009, 83(21): 11090-11101.
[28] Ruggieri A, Dazert E, Metz P, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection[J]. Cell Host Microbe, 2012, 12(1): 71-85.
[29] Garaigorta U, Heim M H, Boyd B, et al. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress[J]. J Virol, 2012, 86(20): 11043-11056.
[30] Raaben M, Groot Koerkamp M J, Rottier P J, et al. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies[J]. Cell Microbiol, 2007, 9(9): 2218-2229.
[31] Sola I, Galan C, Mateos-Gomez P A, et al. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replicationtranscription sites[J]. J Virol, 2011, 85(10): 5136-5149.
[32] Cristea I M, Rozjabek H, Molloy K R, et al. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication[J]. J Virol, 2010, 84(13): 6720-6732.
[33] Frolova E, Gorchakov R, Garmashova N, et al. Formation of nsP3-specific protein complexes during Sindbis virus replication[J]. J Virol, 2006, 80(8): 4122-4134.
[34] Gorchakov R, Garmashova N, Frolova E, et al. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells[J]. J Virol, 2008, 82(20): 10088-10101.
[35] Fros J J, Domeradzka N E, Baggen J, et al. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci[J]. J Virol, 2012, 86(19): 10873-10879.
[36] Mcinerney G M, Kedersha N L, Kaufman R J, et al. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation[J]. Mol Biol Cell, 2005, 16(8): 3753-3763.
[37] Panas M D, Varjak M, Lulla A, et al. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection[J]. Mol Biol Cell, 2012, 23(24): 4701-4712.
[38] Emara M M, Brinton M A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly[J]. Proc Natl Acad Sci U S A, 2007, 104(21): 9041-9046.
[39] Emara M M, Liu H, Davis W G, et al. Mutation of mapped TIA-1/TIAR binding sites in the 3' terminal stem-loop of West Nile virus minus-strand RNA in an infectious clone negatively affects genomic RNA amplification[J]. J Virol, 2008, 82(21): 10657-10670.
[40] Li W, Li Y, Kedersha N, et al. Cell proteins TIA-1 and TIAR interact with the 3' stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication[J]. J Virol, 2002, 76(23): 11989-12000.
[41] Pager C T, Schutz S, Abraham T M, et al. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules[J]. Virology, 2013, 435(2): 472-484.
[42] White J P, Cardenas A M, Marissen W E, et al. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase[J]. Cell Host Microbe, 2007, 2(5): 295-305.
[43] White J P, Reineke L C, Lloyd R E. Poliovirus switches to an eIF2-independent mode of translation during infection[J]. J Virol, 2011, 85(17): 8884-8893.
[44] Borghese F, Michiels T. The leader protein of cardioviruses inhibits stress granule assembly[J]. J Virol, 2011, 85(18): 9614-9622.
[45] Khong A, Jan E. Modulation of stress granules and P bodies during dicistrovirus infection[J]. J Virol, 2011, 85(4): 1439-1451.
[46] Yang X, Nath A, Opperman M J, et al. The doublestranded RNA-dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells[J]. Mol Biol Cell, 2010, 21(19): 3449-3458.
[47] Lu B, Nakamura T, Inouye K, et al. Novel role of PKR ininflammasome activation and HMGB1 release[J]. Nature, 2012, 488(7413): 670-674.
[48] Taghavi N, Samuel C E. Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection[J]. Virology, 2012, 427(2): 208-216.
[49] Steele L, Errington F, Prestwich R, et al. Proinflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming[J]. Mol Cancer, 2011, 10: 20.
[50] Garcia M A, Gil J, Ventoso I, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action[J]. Microbiol Mol Biol Rev, 2006, 70(4): 1032-1060.
REGULATION OF STRESS GRANULES DURING RNA VIRUS INFECTION
WANG Xiao-xu1, SUN Ying-jie2, DING Yun-lei3, WANG Gui-jun1, DING Chan2
(1. College of Animal Science &Technology, Anhui Agricultural University, Hefei 223006, China; 2. Shanghai Veterinary Research Institute, CAAS, Shanghai 200241, China; 3. College of Veterinary Medicine, Nanjing Agricultural University, Nanjing210095, China)
Stress granules (SGs) are reversible dynamic structures that rapidly form when mammalian cells encounter environmental stress like heat shock, oxidative, nutrient deprivation, or viral infection, which can reduce global translation rates. As the complexes of stalled translation factor, SGs play major roles in gene expression and homeostasis. When virus infects host cells, the formation of SGs can greatly reduce the level of protein translation, thereby inhibit viral replication. Yet simple viruses have evolved effi cient means to resist the stress responses, some of them even can use the silent transcripts involved in SGs to increase their replication. This review covers the range of interactions between RNA viruses and SGs and discusses the newly described interactions between stress responses and innate immune responses.
Stress granules; RNA virus; regulation system; innate immune
S852.42
:A
1674-6422(2014)04-0072-09
2014-02-19
863计划(2011AA10A209);公益性行业(农业)科研专项(201003012)
王晓旭,女,硕士研究生,预防兽医学专业
丁铲,E-mail: shoveldeen@shvri.ac.cn;王桂军,E-mail:wangguijun@ahau.edu.cn
