人源多巴脱羧酶的表达、纯化以及高通量抑制剂筛选模型的建立
2014-03-25张缘缘邱军强周越洋张庆华
张缘缘,邱军强,周越洋,吴 方,张庆华
(1. 上海海洋大学水产与生命学院 省部共建水产种质资源发掘与利用教育部重点实验室, 上海 201306;2. 上海交通大学 系统生物医学研究院,上海 200240)
帕金森病(Parkinson′s Disease,PD)是一种中老年常见的神经系统退行性疾病,且疾病发生目前呈年轻化趋势[1]。现有的治疗药物和手术方法等都不能根治此疾病[2]。帕金森病病理的重要特征是患者黑质纹状体中多巴胺能神经元(dopaminergic neurons)大量变性丢失并伴有神经递质多巴胺的缺失,现有的药物主要通过提高神经中枢的黑质纹状体通路中多巴胺的含量来维持多巴胺与乙酰胆碱的动态平衡[3-4]。
多巴胺不能透过血脑屏障,故直接口服或者静脉注射不能补充脑内缺少的多巴胺[5]。左旋多巴(L-Dopa)作为多巴胺合成前体可通过氨基酸转运系统透过血脑屏障进入脑内,但其在外周组织中被多巴脱羧酶(DDC)大量地降解,只有约口服用药量1%的左旋多巴能够到达脑内并被多巴胺神经元摄取后转而变为多巴胺发挥替代治疗作用[6]。因此,临床上抑制外周多巴脱羧酶的活性是十分必要的,左旋多巴和多巴脱羧酶抑制剂的联合用药是治疗的常用方法[7-8]。多巴脱羧酶是一种维生素B6依赖酶。不仅可以催化多巴产生多巴胺,色氨酸产生色胺,也能催化5-羟基色氨酸产生5-羟基色氨。目前研究还发现多巴脱羧酶和高血压、抑郁、失眠、厌食等疾病相关。多巴脱羧酶已知的抑制剂主要是其底物多巴的肼类衍生物(hydrazine),其中以甲基多巴肼(Carbidopa)为代表。这类化学物的抑制机理是通过它们的肼基不可逆地与多巴脱羧酶辅酶维生素B6形成共价键,从而抑制其活性。虽然多巴类似物的肼类衍生物已被作为药物在临床上使用,但它们对多巴脱羧酶的特异选择性不好[9]。
稳定血浆中左旋多巴浓度、延长其半衰期来增加脑内纹状体多巴胺含量的药物成为近年来帕金森病药物研究的热点之一[8, 10]。多巴脱羧酶的抑制剂可以通过抑制外周组织中多巴脱羧酶的活力从而降低左旋多巴在外周组织的降解,提高其生物利用率和药学治疗效果。因此发现新型特异的多巴脱羧酶抑制剂具有重要的理论和实际价值。
目前,多巴脱羧酶的测活方法主要是同位素标记、荧光和Elisa等,还没有报道适合高通量筛选抑制剂的方法。本文将拟建立一种高通量筛选多巴脱羧酶特异抑制剂的模型,为帕金森病以及其它疾病的治疗提供新型的药物先导化合物。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株
载体:pET28b实验室保存;DNA:多巴脱羧酶基因(BC008366)购于美国Proteintech公司;磷酸烯醇式丙酮酸羧化酶(PEPC):大肠杆菌基因组克隆获得;菌株:大肠杆菌(BL21)感受态由作者实验室自制。
1.1.2 试剂
PCR纯化、胶回收纯化以及DNA小抽试剂盒购自Axygen生物科技有限公司,KOD-plus neo购自TOYOBO公司,DNA Marker、限制性核酸内切酶(NdeⅠ,HindⅢ)、T4 DNA连接酶等购自宝生物(TAKARA)公司,预染蛋白质Marker购自Invitrogen公司,引物由上海生工合成,甲基多巴(Methyldopa)、牛血清白蛋白(BSA)和烟酰胺腺嘌呤二核苷酸还原态(NADH)购于上海生工;磷酸吡哆醛(PLP)和左旋多巴(L-Dopa)购于Sigma公司;磷酸烯醇式丙酮酸酯盐(PPPA)购于Alfa Aesar公司;β-巯基乙醇和苹果酸脱氢酶(MDH)购于Amresco公司。
1.1.3 仪器
多功能酶标仪购于Biotek公司; 细胞压力破碎仪购于英国Constant systems公司;BIAcore 3000型表面等离子共振(SPR)传感器购于BIAcore公司。
1.2 方法
1.2.1 克隆和表达
1)基因的克隆。根据Genbank所提供的多巴脱羧酶(DDC)、磷酸烯醇式丙酮酸羧化酶(PEPC)基因序列设计引物(P1、P2、P3和P4)。分别在上游引入NdeⅠ酶切位点和保护碱基,下游引入HindⅢ酶切位点和保护碱基并且在DDC的N-端引入His标签(P1、P2),在PEPC的N端和C端分别引入His标签(P3、P4),引物序列为:P1:5′-TATATATAGCTAGCAACGCAAGTGAATTCCGAAGGAGAGGG-3′;P2:5′-AAATTTAAGCTTCTACTCCCTCTCTGCTCGCAGCACG-3′;P3:5′-TATATATAGCTAGCAACGAACAATATTCCGCATTGCG-3′; P4:5′-AAATTTAAGCTTTCAGTGGTGGTGGTGGTGGTGGCCGGTATTACGCATACCTGCCGC-3′
2)表达质粒的构建。将PCR扩增得到的DDC和PEPC基因产物,用NdeⅠ和HindⅢ双酶切后连接到用同样限制性内切酶酶切的质粒载体pET28b上,以获得重组质粒pET28b-DDC、pET28b-PEPC。
3)重组蛋白的诱导表达。分别将重组质粒pET28b-DDC、pET28b-PEPC转化感受态细胞E.coliBL21(DE3),获得DDC和PEPC表达型重组大肠杆菌,挑取平板单菌落,接种于5 mL含50 μg/mL卡那霉素的LB液体培养基,37℃,250 r/min振荡过夜培养。将1 mL上述菌液接种到1 L含50 μg/mL卡那霉素的LB液体培养基, 37℃,250 r/min振荡培养直至菌液OD600=0.8后,加入异丙基硫代半乳糖苷(IPTG)至终浓度为100 μM,16℃过夜培养,离心后收集的菌体用PBS悬浮,细胞压力破碎仪破胞,用12000 r/min离心30 min将菌体碎片去除。
4)重组蛋白的纯化。取上述上清加到Ni-NTA柱中,孵育1 h,分别加入含有5 mM和50 mM咪唑PBS缓冲液洗杂,然后用8 mL的250 mM咪唑的PBS将蛋白洗脱下来,收集分装并保存在-80℃。
5)SDS-PAGE电泳鉴定。经IPTG诱导前后收集的菌液加入SDS凝胶上样缓冲液,95℃加热5 min,13000 r/min离心1 min后,从上清取20 μL在8%的SDS-PAGE进行蛋白质电泳,之后用考马斯亮蓝染色过夜,脱色后摇荡过夜以消除背景,并记录结果。
6)蛋白浓度的测定。蛋白浓度用BCA Protein Assay kit测定。
1.2.2 酶活测定
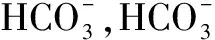
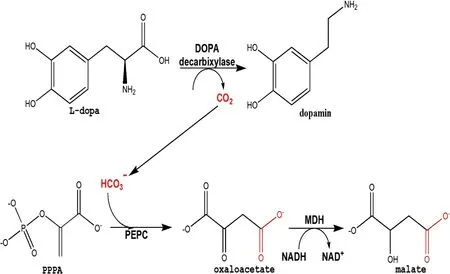
图1 酶联反应原理
2)高通量筛选方法的构建。基于上述酶联反应的测活方法,我们分别检测了不同NADH浓度所对应OD值。同时也测定了时间和pH值对此酶联反应的影响,最终构建了一种高通量筛选多巴脱羧酶的方法。
首先,1 μL的DMSO(对照)或待检测化合物加到384孔板,然后加入24.5 μL酶混合液,包括:65 mM Tris-HCl,65 mM NaCl,0.015%(w/v)BSA,5 mM MgCl2,7.2 mM 巯基乙醇,380 μM NADH,165 nM PEPC,50 μM PLP和142 nM DDC,pH值8.05(终浓度)。随后,加入24.5 μL底物混合液,包括:65 mM Tris-HCl,65 mM NaCl,0.015%(w/v)BSA,5 mM MgCl2,7.2 mM 巯基乙醇,5 mM PPPA,10 U MDH 和0.75 mM L-Dopa(终浓度),pH值8.05。最后用透明封板膜将384孔板封严,立刻用酶标仪读取0 min时数值OD0 min,37℃反应1 h后,再读取60 min数值OD60 min。
计算公式:
剩下活力(%)=
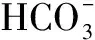
NaHCO3反应模型:类似上述步骤,首先加1 μL的DMSO(对照)或待检测化合物到384孔板,然后加入24.5 μL不含DDC的上述酶混合液,再加入24.5 μL用1.25 μM NaHCO3取代底物L-Dopa的上述底物混合液。
剂量依赖性实验:化合物从200 μM开始倍比稀释7个浓度,其他步骤如上所述。所有数据均独立重复2次,每次实验样品数为3(n=3),实验结果为:平均数±方差。
2 结果与分析
2.1 基因的克隆
a: M—DNA Marker; 1,2—PEPC PCR产物。 b: M—DNA Marker; 1,2—DDC PCR产物。

图3 重组质粒双酶切验证
a: pET28b-DDC双酶切验证(M—DNA marker;1、3—酶切DDC;2、4—未酶切DDC)。b: pET28b-PEPC双酶切验证(M—DNA marker;1、3—酶切PEPC;2、4—未酶切PEPC)。
从大肠杆菌基因组中扩增得到PEPC约2652 bp的基因片段,以及购买的DDC基因扩增得来的约1964 bp的基因片段,PCR扩增后经1%琼脂糖凝胶电泳,结果显示产物片段大小与预期相符(如图2)。通过NdeⅠ和HindⅢ限制性内切酶分别对PCR产物和pET28b载体质粒进行双酶切,再用T4 DNA连接酶将酶切后的PCR产物和pET28b载体连接后,获得重组质粒pET28b-DDC、pET28b-PEPC,经双酶切验证(如图3),分别得到了大小约为1964 bp、2652 bp的目的条带,并且该基因经测序验证,确定目的基因已经成功连接到表达载体上。
2.2 基因在大肠杆菌的表达和纯化
将重组质粒pET28b-PEPC转化感受态细胞E.coliBL21(DE3),获得PEPC表达型重组大肠杆菌BL21/pET28b-PEPC,将重组菌株诱导过夜表达,离心上清再经Ni-NTA柱纯化后,通过SDS-PAGE电泳结果(如图4a)表明,纯化得到了较纯的目的蛋白(约98 kD),并且250 mM咪唑洗脱的PEPC浓度最高(8泳道,图4a)。我们在下面酶活测试实验中使用该洗脱组份的纯酶作为测试反应的偶联酶。我们用相似方法提纯得到了DDC纯酶,且250 mM咪唑洗脱中纯度较高且大小正确(约54 kD,2泳道,图4b),因此选用此提纯后脱盐的组份(1泳道,图4b)去测试DDC的活力。
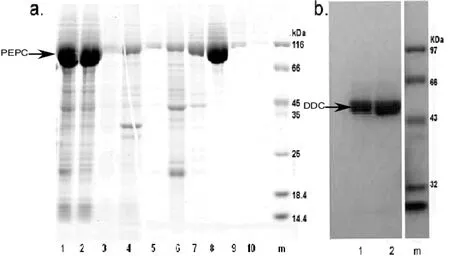
图4 SDS-PAGE电泳分析重组蛋白的原核表达及纯化
a:PEPC的原核表达及纯化(m—蛋白Marker; 1—破包后上清; 2—过柱子流穿; 3—未经诱导的PEPC; 4—诱导后的PEPC; 5—5 mM咪唑PBS缓冲液洗杂; 6—50 mM咪唑PBS缓冲液洗杂; 7—100 mM咪唑PBS缓冲液洗杂; 8—250 mM咪唑PBS缓冲液洗脱; 9—500 mM咪唑PBS缓冲液洗脱; 10—1000 mM咪唑PBS缓冲液洗脱)。b:DDC的原核表达(m—蛋白Marker;1—DDC洗脱组份去盐后;2—250 mM咪唑PBS缓冲液洗脱后的纯化DDC)。

图5 不同浓度NADH对应的OD值
2.3 测定酶活方法
由于NADH在340 nm处有最大吸收峰,而NAD+几乎没有吸收,因此我们检测了不同NADH浓度对应OD值,结果表明OD值和NADH浓度在0.1 mM~1 mM是线性关系(如图5)。为了灵敏的检测NADH的变化,我们选择了NADH浓度约为380 μM、OD值约为1.1的点,作为酶联反应中所用的浓度。
根据L-Dopa与 DDC报道的Km值(~350 μM)[13],我们选取底物L-Dopa浓度为750 μM。检测不同pH值和时间对酶活的影响,优化得到最适反应条件。结果(如图6)表明,0 min时,OD值是1.1。反应一段时间后,OD值下降,下降的值代表此酶促反应的活力(也称窗口)。pH值对酶活影响较为明显,在碱性缓冲液中酶促反应的窗口最大。而酶促反应的时间延长,窗口也相应变大,但会伴有NADH的自身水解。为了减少这一水解现象,我们选择反应时间60 min检测酶活。

图6 在不同pH值缓冲液中,不同时间DDC酶促反应的酶活变化(OD340 nm)Fig 6 The activity of DDC in different pH buffer and different time(OD340 nm)
根据上述反应条件的试验,最终确定了高通量筛选模型,即使用750 μM l-Dopa作为底物,Tris-HCl(pH值8)为反应缓冲液,以380 μM NADH作为检测发光试剂,在反应时间60 min读取测活结果(见实验方法)。 我们用已知的阳性化合物甲基多巴(Methyldopa)测试了这个新型的DDC测活方法[14],结果表明Methyldopa可以抑制DDC的活性,其IC50在1200 μM左右。用1000 μM Methyldopa 做对照,计算此高通量筛选模型的Z-因子的值为~0.55,是在高通量筛选中可接受的范围内(>0.5,通常被认为是合适高通量筛选模型)。
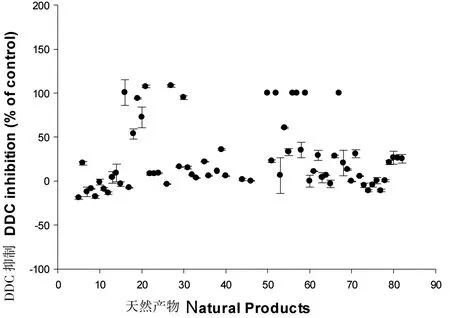
图7 70个天然产物的抑制剂筛选(相对于DMSO的百分比)Fig 7 The high-thoughput screening of 70 natural products (DDC activity,% of DMSO)
2.4 高通量筛选方法的评价
利用建立的筛选方法初筛了70个天然化合物(如图7),主要来自国家小分子药物中心、上海交通大学药学院等单位。其中有11个化合物在200 μM浓度抑制率可以达到90%以上,我们再用这些化合物做复筛实验,从中分析是否在340 nm自身有吸收等干扰因素,并做了剂量依赖性实验,最终确定了2个化合物,即化合物SD-8和SD-48。
我们通过Biacore表面等离子共振仪对这2个化合物进行分析,根据实验结果拟合出SD-8和SD-48的解离常数KD分别为3.4×105和2.6×105,说明这2种化合物对DDC有结合作用。因此可以间接验证我们建立的高通量筛选模型的准确性和科学性。
3 讨论
左旋多巴和多巴脱羧酶抑制剂联合用药的替代疗法已是帕金森病治疗的经典方法。因而发现新的特异有效的小分子抑制剂不仅能提供多巴脱羧酶相关疾病机理研究的特异性探针工具,还能为帕金森病及其他疾病的治疗提供新型的药物先导化合物。
文献报道传统的多巴脱羧酶的测活方法多是用高效液相色谱(HPLC)的方法检测产生底物多巴胺的含量来评价化合物的抑制作用,但筛选出来的抑制剂的特异性不理想;或者用同位素14CO2标记的方法,但这种方法的稳定性不好,因此,这些方法都不适合对该酶进行高通量(HTS)的检测[15-16],荧光、Elisa等方法成本相比较其它方法较高。本模型建立了一种高通量检测CO2的方法,实现了多巴脱羧酶抑制剂高通量的筛选,相比较同位素标记、荧光、Elisa等方法更加快速、微量、准确。
天然产物是自然界生物通过自然选择保留下来的,经历了千百万年的进化过程。它具有结构多样性、化学独特性和类药性。天然化合物比化学合成的化合物更容易形成先导药物结构,可以通过进一步的结构优化得到药物候选化合物,具有较经济的天然产物来源、毒副作用小等特点,是生物活性物质和实用药物发现的重要源泉。人类从大自然植物中发现治疗疾病的药物已经有了长久历史和成功经验。古代“神农尝百草”发现了多种中草药材,近代中国科学家陈克恢从麻黄植物中发现的麻黄碱(一种常用感冒等药物的关键成分之一),美国科学家Wall和Wani从中国南部一种特有的植物喜树(Happy tree)发现的有关抗癌症的药物——喜树碱等[17],这些例子都可以用来证明来源于植物的天然产物在药物研究和发展过程中的重要性。因此。我们建立的多巴脱羧酶抑制剂的高通量筛选模型,以及发现的具有多巴脱羧酶抑制作用的天然产物,可以为多巴胺相关疾病提供药物候选物和疾病研究的小分子探针工具,对和多巴胺相关的疾病研究、治疗和预防具有十分重要的意义。
综上所述,我们建立的多巴脱羧酶抑制剂的高通量筛选模型和发现的具有多巴脱羧酶抑制作用的天然产物可以为帕金森病提供药物候选物和疾病研究的分子探针工具,对和多巴胺相关的疾病治疗和预防具有重要意义。
参考文献:
[1]Morris M E, Martin C L, Schenkman M L. Striding out with Parkinson disease: evidence-based physical therapy for gait disorders[J]. Physical Therapy, 2010, 90(2):280-288.
[2]Rascol O, Payoux P, Ory F, et al. Limitations of current Parkinson's disease therapy[J]. Annals of Neurology, 2003, 53(S3). 3-15.
[3]Hauser R A. Levodopa: past, present, and future[J]. European Neurology, 2008, 62(1): 1-8.
[4]Shulman J M, De Jager P L, Feany M B. Parkinson's disease: genetics and pathogenesis[J]. Annual Review of Pathology: Mechanisms of Disease, 2011(6): 193-222.
[5]Pardridge W M. Blood-brain barrier drug targeting: the future of brain drug development[J]. Molecular Interventions, 2003, 3(2): 90.
[6]Dingemanse J. Issues important for rational COMT inhibition[J]. Neurology, 1999, 55(11 Suppl 4):24-27.
[7]Abdel-Salam O M E. Drugs used to treat Parkinsons disease, present status and future directions[J]. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets, 2008, 7(4):321-342.
[8]Obeso J A, Rodriguez-Oroz M C, Goetz C G, et al. Missing pieces in the Parkinson's disease puzzle[J]. Nature Medicine, 2010, 16(6): 653-661.
[9]Obeso J A, Olanow C W, Nutt J G. Levodopa motor complications in Parkinson's disease[J]. Trends in Neurosciences, 2000, 23:2-7.
[10]Nutt J G, Obeso J A, Stocchi F. Continuous dopamine-receptor stimulation in advanced Parkinson's disease[J]. Trends in NeuroSciences, 2000, 23:109-115.
[11]Smithson D C, Lee J, Shelat A A, et al. Discovery of potent and selective inhibitors of Trypanosoma brucei ornithine decarboxylase[J]. Journal of Biological Chemistry, 2010, 285(22):16771-16781.
[12]Smithson D C, Shelat A A, Baldwin J, et al. Optimization of a non-radioactive high-throughput assay for decarboxylase enzymes[J]. Assay and Drug Development Technologies, 2010, 8(2): 175-185.
[13]Allen G F, Neergheen V, Oppenheim M, et al. Pyridoxal 5′‐phosphate deficiency causes a loss of aromatic l-amino acid decarboxylase in patients and human neuroblastoma cells, implications for aromatic l-amino acid decarboxylase and vitamin B6 deficiency states[J]. Journal of Neurochemistry, 2010, 114(1): 87-96.
[14]Bowsher R R, Henry D P. Aromatic L-amino acid decarboxylase[M]. Neurotransmitter Enzymes, Springer, 1986: 33-78.
[15]Bitonti A J, McCANN P P, Sjoerdsma A. Restriction of bacterial growth by inhibition of polyamine biosynthesis by using monofluoromethylornithine, difluoromethylarginine and dicyclohexylammonium sulphate[J]. Biochem J, 1982, 208: 435-441.
[16]Thyssen S M, Libertun C. Quantitation of polyamines in hypothalamus and pituitary of female and male developing rats[J]. Neuroscience Letters, 2002, 323(1): 65-69.
[17]Oberlies N H, Flora S, Weaver A L. Camptothecin and Taxol: The Story behind the science[J]. Chemistry International, 2003, 25(4): 4-6.
