反复黄疸、上腹痛伴肾病综合征
2014-03-21娄丽璇刘志红
周 岩 娄丽璇 程 震 刘志红
病例摘要
现病史33岁男性,因“反复上腹胀痛、黄疸1年余,蛋白尿4月”于2014-04-21入院。
患者2012年9月出现右上腹胀痛,皮肤巩膜黄染,伴瘙痒、恶心,查直接胆红素149.4 μmol/L,间接胆红素178.4 μmol/L,转氨酶及淀粉酶正常,磁共振示“胆总管胰腺段炎性梗阻可能;胰腺增大、饱满,以胰头部尤甚,增强扫描未见异常强化,DWI见胰头部信号增高;胆囊炎”。行经十二指肠镜逆行胆管造影(ERCP)+鼻胆管引流术,术中见“胆总管下段狭窄,网篮拖出较多小的泥沙样结石,将鼻胆管置入肝总管”,胆总管下段活检病理示“少量黏膜慢性炎症”。对症治疗后复查直接胆红素30.3 μmol/L,间接胆红素44.9 μmol/L,腹痛好转、黄疸减轻,予拔除鼻胆管。2013年5月上述症状再发,磁共振胰胆管成像(MRCP)示“胆总管上段扩张,胰腺体积明显增大,胰头增大,伴斑片状异常强化考虑炎性改变,胆总管下段狭窄,胆囊炎”。腹部CT示腹膜后多发小淋巴结,行剖腹探查+胰腺穿刺活检+胆肠吻合+胆囊切除术,术中见胰腺弥漫肿大,病理示胰腺慢性炎细胞浸润,胰头穿刺及胆囊病理示胰腺组织慢性炎症细胞浸润,慢性胆囊炎急性发作,诊断“胆总管下段狭窄;胆囊炎;自身免疫性胰腺炎”。期间尿检未见明显异常,血清白蛋白(Alb)30.4 g/L。出院后未服药,黄疸、腹痛消失,3月后复查腹部CT示“胰腺缩小”。
2013年6月出现颜面部及双下肢轻度水肿,尿液泡沫增多,查Alb 27 g/L,予输注白蛋白等。12月全身重度水肿,查Alb 11.7 g/L,尿蛋白定量4.15 g/24h,血清肌酐(SCr)122 μmol/L,血IgG 42.8 g/L,IgG4 5 263 mg/L(正常值10~1 350 mg/L),IgA 1.91 g/L,IgM 0.74 g/L,IgE 4 210 IU/mL,免疫固定电泳无异常,补体正常,抗核抗体(ANA)阴性,血常规、肝功能、胆红素正常,自身免疫性肝炎相关抗体均阴性。2014年2月外院肾活检示肾小球基膜弥漫轻度增厚伴节段性钉突形成,上皮下大量嗜复红蛋白沉积;系膜细胞及基质节段性轻度增生;肾小管上皮细胞轻度萎缩;间质纤维化+,少量淋巴-单核细胞浸润,血管无明显改变。免疫荧光:IgG ++++、C3 ++++、C1q ++,颗粒样沉积于基膜,诊断“膜性肾病Ⅰ~Ⅱ期”,予甲泼尼龙40 mg/d治疗半月,静脉滴注环磷酰胺(CTX) 0.4g×1次,同时予利尿、抗感染及输注白蛋白治疗,Alb升至22 g/L。出院后予甲泼尼龙36 mg/d,CTX 0.4g/半月×4次。3月20日予环孢素A(CsA)150 mg/d联合吗替麦考酚酯(MMF) 1 g/d治疗。4月1日我院查尿蛋白定量6.57 g/24h,Alb 26 g/L,SCr 55.7 μmol/L,为进一步诊治收住入院。病程中无消瘦、无颌下及头颈部肿块,无脱发、关节痛、面部红斑、口腔溃疡等。尿量700~1 000 ml/d。
既往史哮喘病史30余年,发作时伴呼气性呼吸困难、咳嗽、咳痰,吸入沙美特罗后数分钟即可缓解,一般10天后可完全缓解。现每日吸入沙美特罗控制,近10年未再复发。2012年8月血压升高,最高175/120 mmHg,服贝那普利及美托洛尔血压控制在110/80 mmHg。
家族史父母均有高血压,父亲有“脑梗塞”史,母亲因“肠癌“去世,2个姐姐均体健,家族中无传染病史。
体格检查血压112/68 mmHg,柯兴貌,面部痤疮,全身皮肤无黄疸。全身未见皮疹及皮下出血点,浅表淋巴结未触及肿大。心律齐,各瓣膜区未闻及病理性杂音,肺部听诊清音,未闻及干湿性啰音。腹软,无压痛,未触及包块,移动性浊音阴性,双下肢轻度水肿。
实验室检查
尿液尿蛋白定量6.89 g/24h,尿沉渣红细胞计数1万/ml,尿白细胞0~1/HP;α2巨球蛋白5 mg/L,C34 mg/L,视黄醇结合蛋白0.3 mg/L,NAG 58.6 U/(g·Cr),尿渗量738 mOsm/(kg·H2O),尿糖阴性。
血液
血常规血红蛋白(Hb) 137 g/L,白细胞计数12.4×109/L,N/L 85.5/9.6,嗜酸细胞比例0.1%,血小板135×109/L。
血生化Alb 26.3 g/L,球蛋白19.4 g/L,血清尿素氮(BUN)7.8 mol/L,SCr 47.7 μmol/L,尿酸321 μmol/L,胱抑素C 1.15 mg/L,谷丙转氨酶44 U/L,谷草转氨酶18 U/L,乳酸脱氢酶503 U/L,总胆固醇7.45 mmol/L,三酰甘油6.26 mmol/L,电解质正常。估算的肾小球滤过率(eGFR)138 ml/(min·1.73m2)(CKD-EPI公式)。空腹血糖 7.5 mmol/L,餐后2h血糖11.5 mmol/L,HbA1c 6.3%。
肿瘤标志物糖类抗原125 41.90 IU/ml、CA72-4 45.37 U/ml,其余标志物正常。
甲状腺功能游离三碘甲腺原氨酸(FT3)4.32 pmol/L,其余正常。
免疫学ANA、A-dsDNA、ENA多肽抗体谱、抗心磷脂抗体、狼疮抗凝因子均阴性。IgG 3.480 g/L,IgA 1.81 g/L,IgM 0.054 g/L,IgE 193 IU/ml,RF阴性,补体正常,外周血淋巴细胞亚群CD4 481个/μl,CD8 300个/μl,CD3 806个/μl,CD20 119个/μl。抗磷脂酶A2受体抗体(A-PLA2R)阴性。两次免疫球蛋白亚类IgG4分别为610 mg/L和1 040 mg/L(30~2 010 mg/L)。
其他乙肝、丙肝标记物阴性。全血CsA谷值浓度103.40 ng/ml(剂量150 mg/d)。
影像学检查
双肾B超左肾:124 mm×55 mm×62 mm,右肾:122 mm×55 mm×61 mm,皮质厚度不清,皮质回声增强,皮髓界限清楚。集合系统:左侧光带分离6 mm,右侧光带分离8 mm。
胸腹CT两肺散在间质性肺炎;左肺下叶后基底段肺炎;双侧胸腔少量积液。轻度脂肪肝;肝左叶小囊肿;胆囊切除及术后改变。
肾活检
光镜15个肾小球中2个球性废弃。余正切肾小球体积增大,肾小球节段系膜区轻度增宽,毛细血管袢开放僵硬,囊壁增厚。PASM-Masson:上皮侧较多嗜复红物沉积(图1A)。肾小管间质慢性病变轻度,少数肾小管上皮细胞刷状缘脱落,间质散在单个核细胞浸润。小动脉平滑肌细胞空泡变性,小叶间动脉弹力层增厚、分层(图1)。
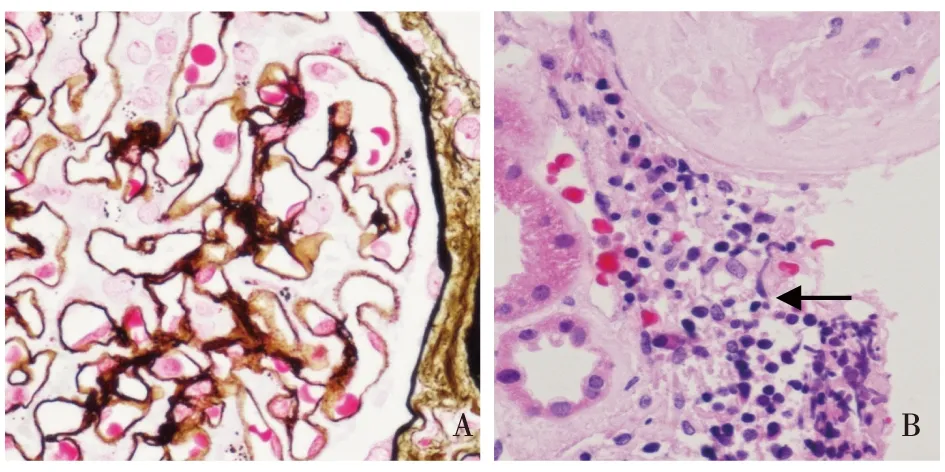
图1 A:肾小球毛细血管袢基膜上皮侧较多嗜复红物沉积(PASM-Mason,×600);B:肾间质少量单个核细胞浸润(↑)(HE,×400)
免疫荧光IgG++、C3+,呈颗粒状弥漫分布于血管袢。IgA、IgM、C1q阴性。IgG 亚型染色 IgG1++、IgG2+、IgG3+、IgG4+++,呈颗粒状弥漫分布于血管袢(图2)。
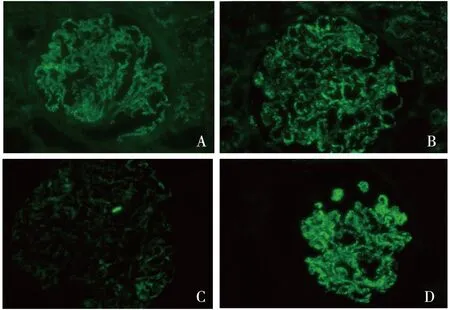
图2 IgG++(A),呈颗粒状弥漫分布于血管袢;IgG1++(B)、IgG3+(C)、IgG4+++(D),呈颗粒状弥漫分布于血管袢(IF,×400)
电镜观察1个肾小球。病变主要在肾小球基膜上皮侧,上皮侧弥漫中~高密度电子致密物沉积,较多电子致密物被钉突分割、包绕;毛细血管袢开放尚好,系膜区、基膜内少量电子致密物沉积。足细胞病变明显,足突广泛融合(80%~90%),胞质大量微绒毛化。肾小管基膜未见电子致密物沉积。
其他肾组织A-PLA2R染色阴性。肾组织IgG4阳性细胞 <50个/HP。
诊断分析青年男性患者,病程1年余,肾脏病的临床表现为肾病综合征,肾活检示肾小球膜性病变,血清及肾组织A-PLA2R阴性,考虑继发性膜性病变,自身抗体、肝炎标志物、肿瘤等检查无异常。肾外尚存在多系统损害:(1)消化系统:反复的梗阻性黄疸伴腹部疼痛,腹部CT示胰腺肿大,曾行剖腹探查术,最后证实不是常见的坏死性胰腺炎或胰腺癌;(2)呼吸系统:幼时有哮喘,现吸入沙美特罗控制;(3)外周血IgG4明显升高。
患者的临床表现需除外IgG4相关性疾病,但外院的胰腺、胆囊活检报告无法证实该诊断。重新行病理切片及IgG4染色发现,胰腺组织的纤维化伴IgG4浆细胞浸润(图3);胆囊IgG4免疫组化染色:IgG4阳性浆细胞染色>50个/HP(图4),证实为自身免疫性胰腺炎和IgG4相关的胆囊炎,最终确诊为IgG4相关疾病。
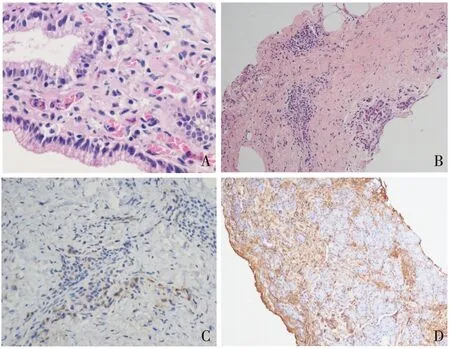
图3 A:胰腺组织少量单个核细胞浸润(HE,×200);B:间质纤维化明显(HE,×100);C、D:IgG4+浆细胞比例少(IgG4染色,C:×400,D:×200)
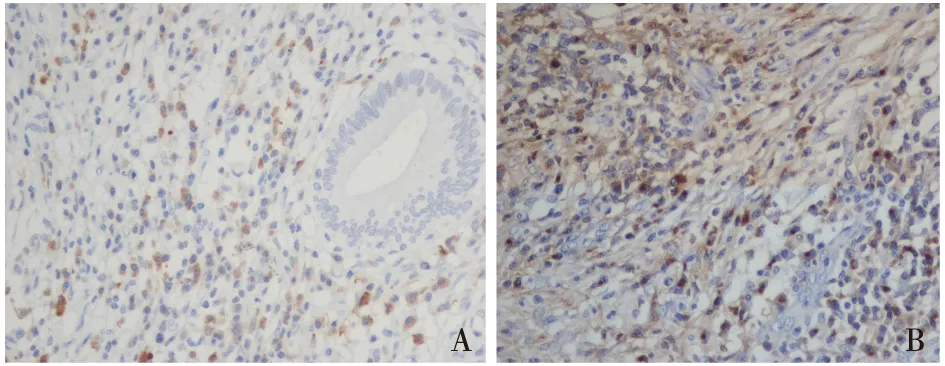
图4 A:胆囊组织IgG4+浆细胞浸润;B:IgG4+浆细胞>50个/HP(IgG4染色,A:×200,B:×400)
治疗经过入院后鉴于患者一般情况差,CsA治疗1月Alb及尿检持续无改善予停药,继续予泼尼松30 mg/d、贝那普利、依折麦布、非诺贝特、阿卡波糖等治疗。
随访6月14日门诊随访尿蛋白定量6.24 g/24h,Alb 18.9 g/L,SCr正常,继续予泼尼松20 mg/d治疗;7月3日复查尿蛋白6.20 g/24h,Alb 24.5 g/L,SCr正常。
最后诊断(1)IgG4相关性疾病;(2)肾小球膜性病变;(3)自身免疫性胰腺炎、胆囊炎;(4)硬化性胆管炎;(5)哮喘;(6)胆囊切除术后。
讨 论
青年男性患者,病程1年余,临床多系统受累(肾脏、胰腺、胆囊、胆管、肺等),外院查血清IgG4水平升高明显。肾脏表现为肾病综合征,肾活检示肾小球膜性病变;消化系统:因反复黄疸及腹痛,CT示胰腺肿大,行手术治疗,病理诊断为自身免疫性胰腺炎及自身免疫性胆囊炎;胆囊组织IgG4阳性浆细胞>50个/HP;呼吸系统表现为哮喘,最终诊断为IgG4相关性疾病(IgG4-RD)。
2003年,Kamisawa等[1]首先提出IgG4-RD,并在2010年正式命名[2],定义为一类可引起多器官纤维化的全身性炎性疾病,几乎可累及所有器官,包括Mikulicz’s病、自身免疫性胰腺炎(AIP)、自身免疫性垂体炎、甲状腺炎、间质性肺炎、间质性肾炎、腹膜后纤维化、炎性主动脉瘤和炎性假瘤。各器官病变可同时或相继出现,我国以胰腺、涎腺及胆道病变为主[3],表现为弥漫性或局灶性器官肿大,伴/不伴血清IgG4升高(>1 350 mg/L),典型组织学特征为淋巴浆细胞浸润、席纹状纤维化、闭塞性静脉炎及IgG阳性浆细胞增多(IgG4占浆细胞总数40%以上),进而导致硬化和纤维化,大部分患者早期应用激素治疗有效,停药后易复发。该病好发于>50岁的男性,起病隐匿,临床表现取决于疾病活动程度和累及器官,主要为局部压迫症状和相应器官功能障碍[4];60%的患者血清IgG4水平可升高,且与受累器官数目呈正相关,但特异度不高。
AIPAIP的临床表现为黄疸(约70%为梗阻性黄疸)、腹痛和体重减轻等。典型CT表现为胰腺弥漫性肿大,呈“腊肠”样,以胰头为主,通常需要内镜下逆行胰胆管造影确诊。根据AIP的诊断标准[5],该患者CT检查提示胰腺弥漫性肿大,血清IgG4大于正常上限的2倍;胰腺组织的纤维化伴IgG4浆细胞浸润,符合IgG4相关的胰腺炎。其胰腺IgG4阳性浆细胞数未达到>10个/HP的标准,可能与取材部位纤维化有关。
IgG4相关硬化性胆管炎(IgG4-SC)IgG4-SC是以胆管壁IgG4阳性细胞浸润和明显纤维化为特征的一种硬化性胆管炎;90%合并AIP;多累及大胆管,表现为梗阻性黄疸,胆管造影可见局灶性或多发性胆管狭窄;类似于壶腹周围癌、胆管癌。本例患者反复黄疸、上腹部疼痛,胰腺肿大,以胰头为主,我院病理科重新将胆囊活检组织染色后提示IgG4相关胆囊炎。结合其梗阻性黄疸、内镜下逆行胰胆管造影、磁共振胰胆管造影均提示胆总管狭窄,虽其病理上未见明显的炎细胞浸润,故考虑存在IgG4-SC。
IgG4相关肾损害IgG4-RD通常表现为急性或慢性肾功能不全及蛋白尿[6],既往认为有以下三种类型:(1)弥漫性间质性肾炎;(2)肾实质外的病变引起的肾脏病变,如腹膜后病变(腹膜后组织纤维化、硬化)引起的肾积水;(3)浆细胞局灶性浸润肾间质引起的肾脏病变。
既往认为,IgG4-RD主要表现为肾小管间质纤维化(TIN),称IgG4相关TIN。近年来,随着IgG4-RD的报道增多[7-9],目前认为其肾小球损害多样(表1)。Watson等[10]于2006年最先报道IgG4相关的膜性肾病(MN),约7%同时存在TIN。Saeki等[6]报道的23例IgG4-TIN中6例合并肾小球病变,2例MN,1例IgA肾病(IgAN),3例为增生性肾小球肾炎伴系膜区或内皮下免疫复合物沉积。AIP合并膜增生性肾炎、IgAN及毛细血管内增生性肾炎的个案报道较多,以合并MN最常见,伴/不伴TIN。目前共报道16例IgG4相关MN的患者,其中9例合并TIN(包括2例局灶性TIN),单纯MN 5例,1例合并IgAN,1例合并局灶内皮增生。多为中老年人(34~83岁),男性13例,女性3例;8例A-PLA2R为阴性,1例阳性,5例未检查,最常见的肾外受损是AIP(50.0%),其次是涎腺炎、淋巴结、肝脏、肺、眼等[8,10-16]。
本例患者的两次肾活检示肾小球膜性病变,A-PLA2R阴性,结合其消化系统损害的临床表现,考虑肾小球膜性病变亦为IgG4相关。
IgG4相关性肺病可分为间质性肺炎和炎性假瘤,无特异性影像学表现,国外回顾性研究报道IgG4-RD的肺受累发生率14%~54%[17]。本例患者幼年时即有哮喘,文献报道IgG4-RD合并哮喘罕见[18];但多数文献认为IgG4-RD以Th2细胞活化为主,常见过敏、嗜酸细胞增多等表现,IgG4-RD和哮喘有共同的发病机制。因无影像学及肺活检病理结果,无法证实肺部疾病是否IgG4相关。

表1 IgG4相关性肾病的诊断标准[19]
激素是治疗IgG4-RD的一线用药,常用初始方案为口服泼尼松龙 0.6 mg/(kg·d)或30~40 mg/d治疗2~4周,在3~6月内减量至5 mg/d。>90%患者激素治疗后临床症状缓解、血清IgG4水平降低及影像学改善[4],疗效取决于受累器官纤维化程度。激素也是治疗复发病例的首选药物,95%患者再次应用仍可获得临床缓解。硫唑嘌呤、MMF及甲氨蝶呤等也可用于维持期治疗。
小结:IgG4-RD是一种累及多器官和组织免疫性疾病,以血清IgG4浆细胞显著增生,进而导致纤维化和硬化为主要特征的慢性疾病。由于累及器官组织广泛,早期临床特征无特殊性,诊断多依靠组织学、影像学、血清学检查,且多个器官组织受累可交叉存在。除表现为TIN外,IgG4相关的肾损害也可表现为肾小球膜性病变,在后者的病因鉴别中应加以关注。
(致谢:胰腺及胆囊组织的病理诊断由南京军区南京总医院病理科李南云教授指导,特此致谢!)
1Kamisawa T,Funata N,Hayashi Y,et al.A new clinicopathological entity of IgG4-related autoimmune disease.J Gastroenterol,2003,38(10):982-984.
2Takahashi H,Yamamoto M,Suzuki C,et al.The birthday of a new syndrome:IgG4-related diseases constitute a clinical entity.Autoimmun Rev,2010,9(9):591-594.
3李坤鹏,朱剑,赵伟,等.IgG4相关性疾病20例临床特征分析.中华风湿病学杂志,2012,16(12):820-824.
4Ebbo M,Daniel L,Pavic M,et al.IgG4-related systemic disease:features and treatment response in a French cohort:results of a multicenter registry.Medicine (Baltimore),2012,91(1):49-56.
5Otsuki M,Chung JB,Okazaki K,et al.Asian diagnostic criteria for autoimmune pancreatitis:consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis.J Gastroenterol,2008,43(6):403-408.
6Saeki T,Nishi S,Imai N,et al.Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis.Kidney Int,2010,78(10):1016-1023.
7Cornell LD,Chicano SL,Deshpande V,et al.Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease.Am J Surg Pathol,2007,31(10):1586-1597.
8Morimoto J,Hasegawa Y,Fukushima H,et al.Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis.Intern Med,2009,48(3):157-162.
9Saeki T,Imai N,Ito T,et al.Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis.Clin Nephrol,2009,71(2):173-178.
10 Watson SJ,Jenkins DA,Bellamy CO.Nephropathy in IgG4-related systemic disease.Am J Surg Pathol,2006,30(11):1472-1477.
11 Alexander MP,Larsen CP,Gibson IW,et al.Membranous glomerulonephritis is a manifestation of IgG4-related disease.Kidney Int,2013,83(3):455-462.
12 Hill P,Russell P,Sammartino C,et al.Acute kidney injury and proteinuria in a patient with diabetes and a submandibular mass.Am J Kidney Dis,2009,54(2):375-380.
13 Cravedi P,Abbate M,Gagliardini E,et al.Membranous nephropathy associated with IgG4-related disease.Am J Kidney Dis,2011,58(2):272-275.
14 Li XL1,Yan TK,Li HF,et al.gG4-related membranous nephropathy with high blood and low urine IgG4/IgG ratio:a case report and review of the literature.Clin Rheumatol.2014 Jan;33(1):145-148.
15 Fervenza FC,Downer G,Beck LH Jr,et al.IgG4-related tubulointerstitial nephritis with membranous nephropathy.Am J Kidney Dis,2011,58(2):320-324.
16 Jindal N,Yadav D,Passero C,et al.Membranous nephropathy:a rare renal manifestation of IgG4-related systemic disease.Clin Nephrol,2012,77(4):321-328.
17 Ryu JH,Sekiguchi H,Yi ES.Pulmonary manifestations of immunoglobulin G4-related sclerosing disease.Eur Respir J,2012,39(1):180-186.
18 Lee YS,Cho HJ,Yoo HS,et al.A case of IgG4-related disease with bronchial asthma and chronic rhinosinusitis in Korea.J Korean Med Sci,2014,29(4):599-603.
19 Kawano M,Saeki T,Nakashima H,et al.Proposal for diagnostic criteria for IgG4-related kidney disease.Clin Exp Nephrol,2011,15(5):615-626.
