HPLC法同时测定甘草中18α-甘草酸和18β-甘草酸的含量Δ
2014-03-09辽宁中医药大学药学院辽宁大连116600
陈 莹,赵 博,乔 佳,张 慧(辽宁中医药大学药学院,辽宁大连 116600)
甘草酸是从豆科植物甘草中提取的一种三萜皂苷,是甘草中最重要的有效成分之一[1]。甘草酸具有两种构型,分别是18α-甘草酸(甘草酸二铵)和18β-甘草酸(甘草酸单铵),二者互为反向异构体,在甘草中同时存在。18α-甘草酸和18β-甘草酸均具有抗炎、保肝、抗纤维化、抗肿瘤等药理作用[2-4],由二者制成的片剂、注射剂在临床上广泛应用[5-6]。目前多见采用高效液相色谱(HPLC)法测定甘草中18β-甘草酸含量的报道[7-9],尚未见测定甘草中18α-甘草酸含量及方法的报道。为了更好地掌控甘草药材的质量,笔者采用HPLC法在同一色谱条件下同时测定甘草中18α-甘草酸和18β-甘草酸的含量,试验结果可为不同商品规格甘草药材的质量评价提供参考。
1 材料
1.1 仪器
LC-10A型HPLC系统,包含紫外检测器、LC solution 色谱工作站等(日本岛津公司);CP225D型电子分析天平(德国Sartorius 公司);KQ5200DB 型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试剂
18α-甘草酸、18β-甘草酸对照品(中国食品药品检定研究院,批号:101050-201101、110731-201116,含量均≥98.0%);乙腈为色谱纯,其余试剂均为分析纯,水为哇哈哈纯净水。
1.3 药材
市售10 个不同商品规格的甘草药材,均经辽宁中医药大学中药鉴定教研室翟延君教授鉴定为豆科植物甘草(Glycyrrhiza uralensis Fisch.)、胀果甘草(Glycyrrhiza inflata Bat.)或光果甘草(Glycyrrhiza glabra L.)的干燥根和根茎,详见表1。

表1 不同商品规格样品的商家和来源Tab 1 Manufacturers and sources of samples with different specifications
2 方法与结果
2.1 色谱条件
色谱柱:Ecosil-C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.1%磷酸盐缓冲液(80 ∶20,V/V,pH 7);流速:1.0 ml/min;检测波长:250 nm;柱温:30 ℃;进样量:10 μl。
2.2 溶液的制备
2.2.1 对照品溶液的制备 分别精密称取18α-甘草酸、18β-甘草酸对照品各适量,置于同一25 ml 量瓶中,加70%乙醇溶解并定容,制成18α-甘草酸、18β-甘草酸的质量浓度均为0.2 mg/ml的混合溶液,作为对照品溶液。
2.2.2 供试品溶液的制备 取甘草药材粉末(过三号筛)约0.2 g,精密称定,置于具塞锥形瓶中,精密加入70%乙醇100 ml,密塞,称定质量,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,再称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液作为供试品溶液。
2.3 系统适用性试验
在“2.1”项色谱条件下,分别取“2.2”项下对照品溶液和供试品溶液10 μl,注入HPLC仪,记录色谱,详见图1。系统适用性试验结果表明,理论板数以18β-甘草酸峰计算为4 400,分离度>1.5。
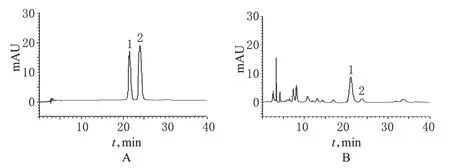
图1 高效液相色谱图A.对照品;B.供试品;1.18β-甘草酸;2.18α-甘草酸Fig 1 HPLC chromatogramsA.substance control;B.test samples;1.18β-glycyrrhizic acid;2.18αglycyrrhizic acid
2.4 线性关系考察
精密称取18α-甘草酸对照品10.14 mg、18β-甘草酸对照品10.28 mg,分别置于同一10 ml量瓶中,用流动相溶解并稀释至刻度,摇匀,作为贮备液。精密量取上述贮备液0.03、0.5、1.0、1.5、3.0 ml,分别置于10 ml 量瓶中,用流动相溶解并稀释至刻度,摇匀,制成系列溶液,分别取10 μl 注入HPLC 仪,记录色谱。以质量浓度(x,μg/ml)为横坐标,峰面积(y)为纵坐标,进行线性回归,得18α-甘草酸和18β-甘草酸回归方程y=4×106x+6 876.1(r=0.999 9)、y=4×106x+2 650.4(r=0.999 9)。结果表明,18α-甘草酸、18β-甘草酸检测质量浓度分别在3.042~304.2、3.084~308.4 μg/ml范围内与各自峰面积积分值呈良好的线性关系。
2.5 精密度试验
精密吸取“2.2.1”项下对照品溶液适量,按“2.1”项下色谱条件重复进样6次,记录各色谱峰面积。结果,18α-甘草酸、18β-甘草酸的RSD分别为0.86%、0.92%,表明仪器的精密度良好。
2.6 稳定性试验
取同一商品规格甘草药材粉末(沈阳成大方圆药店)适量,分别按“2.2.2”项下方法制备供试品溶液,放置0、2、4、6、8 h时按“2.1”项下色谱条件进样,记录各色谱峰面积。结果,18α-甘草酸、18β-甘草酸的RSD分别为0.63%、0.82%,表明供试品溶液在8 h内质量稳定。
2.7 重复性试验
取同一商品规格甘草药材粉末(沈阳成大方圆药店)适量,分别按“2.2.2”项下方法平行制备6 份供试品溶液,并按“2.1”项下色谱条件进样,记录各色谱峰面积,计算含量。结果,含18α-甘草酸、18β-甘草酸的量分别为0.91%、4.38%,RSD分别为0.75%、1.02%,表明本方法的重复性良好。
2.8 加样回收率试验
精密称取0.1 g 已知含量的甘草药材粉末(沈阳成大方圆药店),共6 份,分别精密加入18α-甘草酸、18β-甘草酸对照品溶液适量,按“2.2.2”项下方法制备供试品溶液,并按“2.1”项下色谱条件进样测定,计算加样回收率,结果见表2。

表2 加样回收率试验结果(n=6)Tab 2 Results of recovery test(n=6)
2.9 样品含量测定
取10 个商品规格甘草药材粉末各3 份,精密称定,按照“2.2.2”项下方法制备供试品溶液,并按“2.1”项下色谱条件进样测定峰面积,按外标法计算含量,结果见表3。
表3 样品含量测定结果(%,,n=3)Tab 3 Results of content determination of samples(%,,n=3)

表3 样品含量测定结果(%,,n=3)Tab 3 Results of content determination of samples(%,,n=3)
-:表示未检出-:means no detected
3 讨论
3.1 检测波长的确定
精密称取18α-甘草酸、18β-甘草酸对照品适量,加流动相溶解,分别制成每1 ml约含30 μg的溶液,在200~400 nm波长范围扫描。结果显示,二者均在(250±2)nm波长处有最大吸收,因此选择250 nm作为含量测定的检测波长。
3.2 流动相的确定
笔者曾采用0.05%磷酸溶液-乙腈梯度洗脱的方法[10],结果所需洗脱时间较长;还曾采用甲醇-0.2 mol/L醋酸铵-冰醋酸为流动相[7-8],结果主峰未与杂质峰分离。最终,确定本试验的流动相为乙腈-0.1%磷酸盐缓冲液(80 ∶20,V/V,pH 7)。在此条件下洗脱时间短,且主峰与杂质峰分离度良好。
3.3 样品提取方法的确定
根据18α-甘草酸和18β-甘草酸的溶解性,笔者曾尝试用甲醇、70%乙醇、水进行提取,结果70%乙醇的提取效果最好。笔者还曾尝试采用回流提取法和超声提取法等不同提取方法,结果超声提取法的提取率较高;同时,亦比较了超声20、30、45 min的提取效果,结果超声提取30 min时18α-甘草酸和18β-甘草酸就已提取完全。
3.4 含量测定结果的分析
含量测定结果显示,甘草药材中18β-甘草酸含量最高者可达6.30%,而18α-甘草酸含量最高者仅为0.93%,且有部分样品甚至未检出,可见甘草药材中18α-甘草酸和18β-甘草酸的含量差异较大,18β-甘草酸的含量远高于18α-甘草酸。正因为18β-甘草酸在甘草中的含量较高,因而受到研究者的广泛关注,对另一有效成分18α-甘草酸的研究却往往被忽视。
文献[7-9,11-12]曾报道,采用HPLC 法以甲醇-0.2 mol/L 醋酸铵-冰醋酸、乙腈-0.1 mol/L磷酸溶液为流动相测定甘草中18β-甘草酸含量,以乙腈-2%冰醋酸、甲醇-冰醋酸-水(三乙胺调pH至4.35)为流动相测定甘草制剂中18β-甘草酸含量的方法,但未见对甘草中另一活性成分18α-甘草酸含量测定方法的研究报道,特别是在同一流动相条件下同时检测具有药理活性且为同分异构体的上述两种专属性成分的研究报道。
综上所述,本方法准确、可靠、简单、快速,可用于不同商品规格甘草药材的质量评价。
[1]赵祎镭,师清芝,唐星.甘草中甘草酸和甘草苷的提取纯化工艺研究[J].中国药房,2009,20(6):426.
[2]郑艳,胡汉昆.甘草次酸与甘草酸二铵抗炎护肝作用的实验研究[J].中国药师,2012,15(5):604.
[3]欧强,陈良,谭德明,等.甘草酸二铵抗大鼠免疫性肝纤维化作用研究[J].中成药,2006,28(7):1 046.
[4]刘军花,魏华波,姚瑛,等.四种不同甘草有效成分的抗肿瘤血管新生作用[J].现代生物医学进展,2010,10(14):2 731.
[5]董大勇.异甘草酸镁治疗慢性乙型肝炎的临床观察[J].实用临床医药杂志,2010,14(19):68.
[6]陈凯红,蒋祥虎,蒲云川,等.甘草酸二铵治疗慢性乙型肝炎患者的细胞免疫功能变化[J].中华传染病杂志,2006,24(5):348.
[7]徐树芸,王海宁.HPLC测定甘草提取物中甘草酸的含量[J].安徽农业科学,2008,36(24):10 297.
[8]张崇禧,杨春花,蔡恩博,等.HPLC分析甘草中甘草酸和甘草苷含量[J].资源开发与市场,2010,26(8):673.
[9]牛小宇,刘春生,崔浩然.栽培甘草群体中不同单株甘草酸的含量差异研究[J].时珍国医国药,2009,20(9):2 121.
[10]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:80.
[11]曾英彤,黄志军,李泽华.HPLC 法测定复方甘草片中甘草酸含量[J].中国药师,2009,12(11):1 584.
[12]李浩,刘燕,张少文.高效液相色谱法测定复方甘草合剂中甘草酸含量[J].中国医院药学杂志,2004,24(5):314.
