HPLC 法测定复方吡拉西坦脑蛋白水解物片中硫酸软骨素钠的含量
2014-03-09刘志辉梅州市食品药品监督检验所广东梅州514071
刘志辉(梅州市食品药品监督检验所,广东梅州 514071)
复方吡拉西坦脑蛋白水解物片为复方制剂,主要成分为吡拉西坦、脑蛋白水解物、谷氨酸、硫酸软骨素钠、维生素B1、维生素B2、维生素B6、维生素E;适用于急慢性脑血管病、脑外伤、各种中毒性脑病等原因引起的脑神经功能障碍及记忆力减退、先天性脑发育不全、儿童智能发育迟缓、老年性痴呆等的治疗[1]。其中硫酸软骨素钠主要为N-乙酰半乳糖胺(2-乙酰胺-2-脱氧-β-D-吡喃半乳糖)和D-葡萄糖醛酸的共聚物的硫酸酯钠盐,共聚物内己糖通过β-1,3及β-1,4糖苷键交替连接。复方吡拉西坦脑蛋白水解物片中硫酸软骨素钠测定方法现行标准是分光光度法[2]:利用盐酸水解供试品生成氨基己糖,然后在碱性条件下与乙酰丙酮反应,生成色原物质,再与对二甲氨基苯甲醛溶液反应产生红色,以氨基葡萄糖为对照品,用分光光度法在525 nm 波长处测吸光度,乘以系数0.830 9 计算氨基葡萄糖的量。该法水解产生的氨基己糖系一种氨基半乳糖,半乳糖是一种由6个碳和1个醛组成的单糖,归类为醛糖和己糖;葡萄糖是己醛糖,氨基半乳糖与氨基葡萄糖的分子式相同,但结构式是完全不同的,因此该分光光度法不仅不能准确测定硫酸软骨素钠的主体结构的真实含量,而且结果重现性不好[3]。笔者参考文献[4-8],建立了采用高效液相色谱(HPLC)法测定复方吡拉西坦脑蛋白水解物片中硫酸软骨素钠含量的方法,结果表明该方法灵敏度高、专属性强、操作简单、结果准确,可为控制该药品的质量提供参考。
1 材料
1.1 仪器
1200型HPLC仪、紫外检测器(美国Agilent公司)。
1.2 药品与试剂
硫酸软骨素钠对照品(中国食品药品检定研究院,批号:140792-201001,纯度:100%);样品为复方吡拉西坦脑蛋白水解物片(复方制剂,白求恩医科大学制药厂二分厂,批号:20130620、20121013、20131102,规格:每片含硫酸软骨素钠20 mg);磷酸二氢铵为分析纯;水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:JADE-PAK ODS-AQ(250 mm×4.6 mm,5 μm);流动相:0.008 mol/L磷酸二氢铵溶液(pH=3.6),流速:0.5 ml/min;紫外检测器,检测波长:194 nm;柱温:30 ℃;进样量:10 μl。
2.2 溶液的制备
2.2.1 对照品溶液。精密称取在105 ℃干燥4 h的硫酸软骨素钠对照品约25 mg,置于25 ml 量瓶中,加水溶解并稀释至刻度,摇匀;精密量取5 ml,置于200 ml 量瓶中,加水稀释至刻度,摇匀,作为对照品溶液。
2.2.2 供试品溶液。取样品20 片,精密称定,研细,精密称取适量(约相当于硫酸软骨素钠25 mg),置于50 ml量瓶中,加乙腈5 ml,加水25 ml,超声(功率:250 W,频率:45 kHz)约10 min使硫酸软骨素钠溶解,加水稀释至刻度,摇匀,滤过;精密量取续滤液10 ml,置于200 ml量瓶中,加水稀释至刻度,作为供试品溶液。
2.2.3 阴性样品溶液。按样品处方比例,制备除硫酸软骨素钠外的阴性样品,并按“2.2.2”项下方法制备阴性样品溶液。
2.3 系统适用性试验
取供试品(批号:20130620)溶液注入色谱仪,记录色谱图,以硫酸软骨素钠峰计理论板数为11 369,硫酸软骨素钠峰与相邻峰的分离度为2.01,见图1。
2.4 专属性试验
分别吸取供试品溶液、对照品溶液和阴性样品溶液,注入色谱仪。结果,阴性样品溶液在与对照品相应的保留时间处,无色谱峰出现,表明处方中各成分及辅料不干扰硫酸软骨素钠的含量测定,见图1。
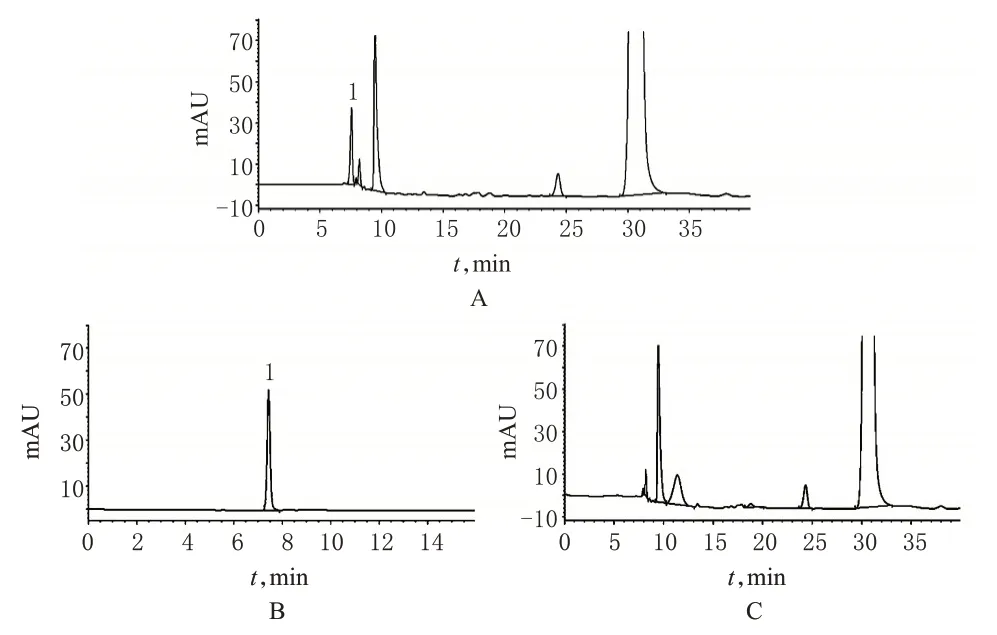
图1 系统适用性试验高效液相色谱图A.供试品溶液;B.对照品溶液;C.阴性样品溶液;1.硫酸软骨素Fig 1 HPLC chromatograms of system suitability testA.sample solution;B.reference solution;C.negative solution;1.chondroitin sulfate
2.5 破坏性试验
分别取样品细粉(批号:20130620)适量(约相当于硫酸软骨素钠25 mg),置于50 ml量瓶中,其中3份分别加入0.1 mol/L盐酸溶液5 ml、0.1 mol/L 氢氧化钠溶液5 ml 和30%双氧水2 ml,放置30 min后中和,加乙腈5 ml、水25 ml,超声(功率:250 W,频率:45 kHz)约10 min,加水稀释至刻度,摇匀;另外2 份分别置于3 000 lx光线下放置6 h和100 ℃加热12 h后,加乙腈5 ml、水25 ml,超声约10 min,加水稀释至刻度,摇匀。取上述溶液注入色谱仪。结果显示,硫酸软骨素钠对酸、碱和光照均较稳定,杂质未见明显变化,加热条件下发生降解,但在氧化条件下更易降解;各破坏条件下的降解产物峰对硫酸软骨素钠峰均无干扰,分离效果良好,见图2。
2.6 定量限和检测限试验
取对照品溶液,以不同比例稀释,在上述色谱条件下测定。以信噪比为10∶1确定定量限为0.75 μg/ml,即7.5 ng;以信噪比为3∶1确定检测限为0.25 μg/ml,即2.5 ng。
2.7 线性关系考察
精密称取硫酸软骨素钠对照品约62.5 mg,置于50 ml 量瓶中,加水振摇使溶解并稀释至刻度,摇匀,作为对照品贮备液。分别精密量取对照品贮备液1、1.5、1、2、3、1、5 ml,置于250、250、100、100、100、25、100 ml 量瓶中,用水稀释至刻度,制备成5、7.5、12.5、25、37.5、50、62.5 μg/ml 的硫酸软骨素钠对照品系列溶液,按上述色谱条件进样测定,记录色谱图,以峰面积(A)为纵坐标、对照品质量浓度(c)为横坐标,绘制标准曲线,得线性方程A=19 166.284 6c+4.589 3(r=0.999 9)。结果表明,硫酸软骨素钠检测质量浓度线性范围为5~62.5 μg/ml。
2.8 精密度试验
取“2.12”项下对照品溶液,连续进样6 次,其峰面积RSD为0.1%(n=6)。
2.9 重复性试验
取同一批号的样品,按“2.2.2”项下方法处理制备、进样,平行测定6 次,平均含量为19.66 mg,RSD 为0.3%(n=6)。
2.10 回收率试验
取已知含量的样品(批号:20130620,每片含19.52 mg),加入硫酸软骨素钠对照品分别制备成相当于标示量80%、100%、120%含量的样品,各3个共9份样品,按含量测定方法测定含量,结果平均回收率为99.1%,RSD=0.7%。结果见表1。

图2 破坏性试验高效液相色谱图A.酸破坏后样品;B.碱破坏后样品;C.光照破坏后样品;D.热破坏后样品;E.氧化破坏后样品;1.硫酸软骨素Fig 2 HPLC chromatograms of destruction testA.sample destructed by acid;B.sample destructed by alkali;C.sample destructed by light;D.sample destructed by high temperature;E.sample destructed by oxidation;1.chondroitin sulfate
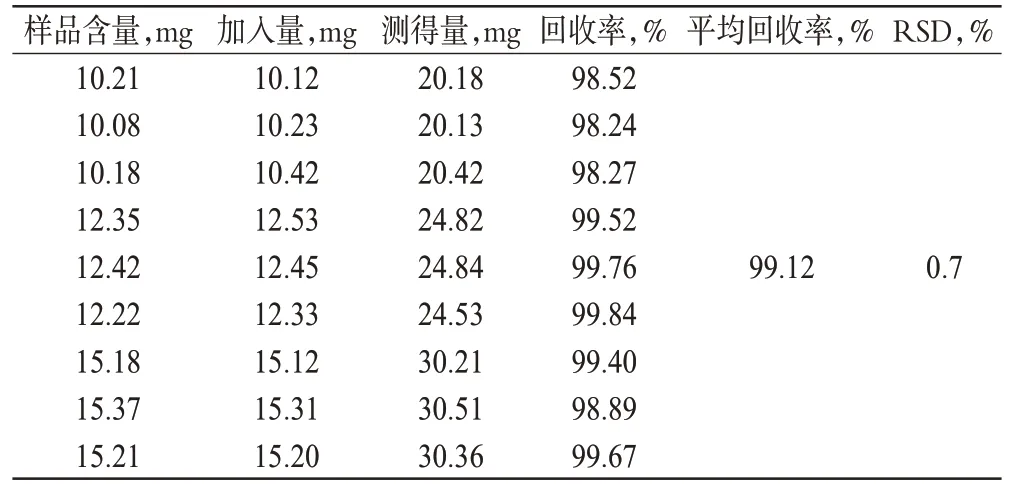
表1 回收率试验(n=3)Tab 1 Results of recovery tests(n=3)
2.11 稳定性试验
取同一份供试品(批号:20130620)溶液,在室温下放置0、2、4、6、8、12 h后测定,结果硫酸软骨素钠的峰面积的RSD=0.6%(n=6)。结果表明,供试品溶液在12 h内稳定。
2.12 样品含量测定
按“2.2.1”“2.2.2”项下方法制备供试品溶液和对照品溶液,按“2.1”项下的色谱条件测定,外标法计算含量,分别对3批样品进行含量测定,结果见表2。

表2 样品含量测定结果Tab 2 Results of content determination of samples
3 讨论
3.1 检测波长的选择
取对照品溶液,于190~400 nm 波长范围内扫描绘制其紫外吸收光谱,结果,硫酸软骨素钠在194 nm 波长处有最大吸收,故本次试验选其为检测波长。
3.2 色谱条件的选择
3.2.1 流动相的选择。分别试用辛烷对氨基苯磺酸钠溶液、庚烷磺酸钠溶液[4-5]和磷酸二氢铵溶液作为流动相,结果发现峰形均好,出峰时间分别为5.957、4.854、7.546 min,与相邻峰的分离度分别为1.36、1.24、2.01,但前2 种溶液为流动相时出峰太快,且分离效果不太好,因此最终采用磷酸二氢铵溶液。因一般情况下加入乙腈能增加洗脱能力,使峰形更好,故笔者尝试加入一定比例的乙腈[4-5]:0.008 mol/L磷酸二氢铵溶液-乙腈(97 ∶3)、0.008 mol/L 磷酸二氢铵溶液-乙腈(95 ∶5)、0.008 mol/L磷酸二氢铵溶液-乙腈(90 ∶10),结果并未见柱效提高和分离效果更好,同时出峰时间却加快了不少,故未加入乙腈。也尝试过选用不同离子强度的磷酸二氢铵溶液(0.008、0.010、0.015 mol/L),结果发现不同离子强度下柱效和分离效果未见明显变化,因此选用0.008 mol/L 的离子强度。色谱柱选用了水性柱,其不但能够支持100%的无机相,且不会损害色谱柱。
3.2.2 溶剂的选择。样品为复方制剂,其中吡拉西坦、维生素B1、维生素B6、硫酸软骨素钠溶于水,谷氨酸微溶于水,脑蛋白水解物为蛋白质类。当以水作溶剂时,在硫酸软骨素峰附近出现有较大的峰,与硫酸软骨素峰层叠在一起分不开;当加入5 ml 乙腈作为提取溶剂时,因减少了其他水溶性物质的溶解度,结果表明能较好地分开硫酸软骨素峰与相邻峰。因此采用25 ml水中加入5 ml乙腈为提取溶剂。
复方吡拉西坦脑蛋白水解物片是复方制剂,成分比较多,若参照文献进行酶解[3],硫酸软骨素可酶解成硫酸软骨素A、B、C,而硫酸软骨素A、B、C 与相邻峰难以分开。本试验则不进行酶解试验,故直接测定的成分是硫酸软骨素钠,操作较为简单,故可作为测定硫酸软骨素钠含量的方法依据。
[1]白求恩医科大学制药厂二分厂.复方吡拉西坦脑蛋白水解物片说明书[S].2010.
[2]国家药典委员会.国家药品标准:化学药品地方标准上升国家标准:第十六册[S].2003:317-318.
[3]任丽萍,于海洲,宋玉娟,等.酶解液相色谱法测定硫酸软骨素钠含量[J].药物分析杂志,2012,32(7):1 246.
[4]狄平平,劳苑子,赵立平,等.HPLC 法测定复方氨基葡萄糖片中盐酸氨基葡萄糖和硫酸软骨素含量[J].中国药事,2010,24(3):283.
[5]石瑞平,胡海勋.离子对反相高效液相色谱法测定硫酸软骨素滴眼液的含量及有关物质[J].药物分析杂志,2008,28(9):1 574.
[6]金鹏飞,吴学军,邹定.HPLC-DAD 同时测定复方尿维氨滴眼液中3 种活性成分的含量[J].中国药学杂志,2009,44(21):1 669.
[7]牛增元,袁玲玲,叶曦雯.高效液相色谱法同时测定硫酸软骨素和盐酸氨基葡萄糖的含量[J].药物分析杂志,2006,26(6):802.
[8]曾芬.RP-HPLC 法测定鲨芪康胶囊中硫酸软骨素含量[J].海峡药学,2010,22(7):88.
