甲基苯丙胺光谱性质的密度泛函分析与指认
2014-03-02
(浙江省台州学院医药化工学院, 浙江 台州316000)
甲基苯丙胺光谱性质的密度泛函分析与指认
陈凯浩,钟爱国
(浙江省台州学院医药化工学院, 浙江 台州316000)
采用密度泛函理论的DFT/B3LYP/6-311+G(d,p)方法和基组, 对甲基苯丙胺的UV-Vis光谱,IR光谱, 1HNMR光谱和荧光光谱进行了理论模拟和指认。自然电荷计算表明,胺基N和H原子很可能是其发挥药理活性的亲电和亲核反应中心。
冰毒;密度泛函理论;电子光谱
甲基苯丙胺(冰毒, C10H15N)是一种人工合成的中枢神经兴奋剂,是我国规定管制的精神药品。它已逐步取代本世纪流行的鸦片、海洛因、大麻、冰毒、可卡因等常用毒品,成为21世纪全球范围滥用最为广泛的毒品。1919年日本化学家A. Ogata将麻黄碱与红磷及碘还原,首次合成了甲基苯丙胺,并用于治疗哮喘和鼻炎。近年境外传入的“摇头丸”,也是冰毒的衍生物。赵金涛等[1]对冰毒的拉曼散射振动模式进行了研究;姚红艳等[2]对盐酸甲基苯丙胺的核磁共振(NMR)等光谱进行了实验测定;张金庄[3]利用红外显微镜,直接采集塑料自封袋表面微量毒品的红外光谱图,并与毒品标准红外光谱的吸收峰尤其是特征吸收峰进行定性比对分析。郑天等[4]建立了应用荧光分光光度法测定缴获冰毒中甲基苯丙胺的含量的方法。张润生等[5]采用气相色谱-傅立叶变换红外光谱联用技术,建立了9种苯丙胺类毒品及其衍生物的分析鉴别方法。由于毒物分析结果常作为法庭评判依据,所以对分析结果的评定需全面的图谱数据作为支撑材料。本文采用密度泛函理论方法, 对甲基苯丙胺的分子光谱如紫外-可见吸收,红外吸收,拉曼吸收,核磁共振吸收以及荧光发射光谱等进行了理论模拟和指认,取得了与实验相吻合的结果。
1 计算方法
本文对冰毒分子采用DFT理论的B3LYP方法在6-311+G(d,p)基组水平上进行了优化计算。在优化得到的稳定构型基础上,采用Freq方法进行了频率分析,结果表明所有简谐振动频率全部为正值,表明其计算结果是可信的。本项目的全部计算工作通过Gaussian 03程序包在PC机上完成(图1)。

图 1 甲基苯丙胺(C10H15N)分子结构和原子编号Fig.1 C10H15N molecular structure and atomic number
2 结果与讨论
2.1 紫外吸收光谱
采用TD DFT/B3LYP/6-311+G(d,p)//DFT/B3LY P/6-311+G(d,p)方法,模拟显示(图2),甲基苯丙胺分
子在258 nm (强吸收)和196 nm(弱吸收)处显示了紫外吸收峰,前者可归属于电子从最高占据轨道(H OMO)跃迁到最低空轨道(LUMO),后者则可归属于电子从次高占据轨道(HOMO-1)跃迁到最低空轨道(LUMO)。这与其标准图谱显示甲基苯丙胺分子在2 60 nm处有较强的紫外吸收峰相吻合[6]。
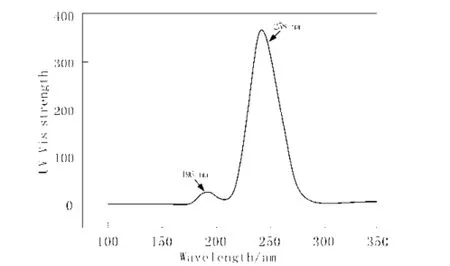
图2 模拟甲基苯丙胺分子的紫外吸收光谱Fig. 2 Simulation UV absorption spectrum of methamphetamine
2.2 拉曼吸收光谱
拉曼效应起源于分子振动与转动, 因此从拉曼光谱中可以得到与红外分子振动互补的信息。采用密度泛函理论DFT/B3LYP方法,在6-311+G(d,p)基组水平上进行了分子构型优化和频率分析,得到6个特征峰。对6个特征峰的分子振动模式进行了详细的归属指认。甲基苯丙胺由异丙基和甲胺构成的支链,取代了苯环上的一个氢原子,形成单取代苯类化合物。图 4是模拟的甲基苯丙胺分子的拉曼散射谱特征峰,主要位于3 450 cm-1(CH3和CH2对称、反对称谱线), 3 190 cm-1(单取代苯有5个C-H 伸缩振动)和1 380 cm-1(CH3和CH2基变形形式的倍频或合频), 1 590 cm-1(面内 C-H 变形衍生的苯环的振动)以及690 cm-1(内环变形), 440 cm-1(三角形环呼吸振动)范围内。对比实验拉曼光谱和理论拉曼光谱,它们的吸收峰基本吻合[7]。
2.3 红外吸收光谱
物质的红外光谱是其分子结构的反映,谱图中的吸收峰与分子中各基团的振动形式相对应(图3)。采用密度泛函理论方法DFT/B3LYP/6-311+G(d, p)模拟显示, 冰毒分子在440 cm-1(弱,顺式构型),690 cm-1(强,反式构型),1 380 cm-1(弱,甲基的C -H对称弯曲振动),1 590 cm-1(强,苯环的C-H面外变形振动吸收峰) ,3 190 cm-1(中强,N-H伸缩振动),3 450 cm-1(弱,C-H基的伸缩振动)等处分别显示了红外吸收峰,这些峰与冰毒分子实验IR图谱在698.4 cm-1, 1 328.7cm-1, 1 570.7cm-1, 2 93 3.4 cm-1,3 500.8 cm-1有吸收基本吻合[5]。
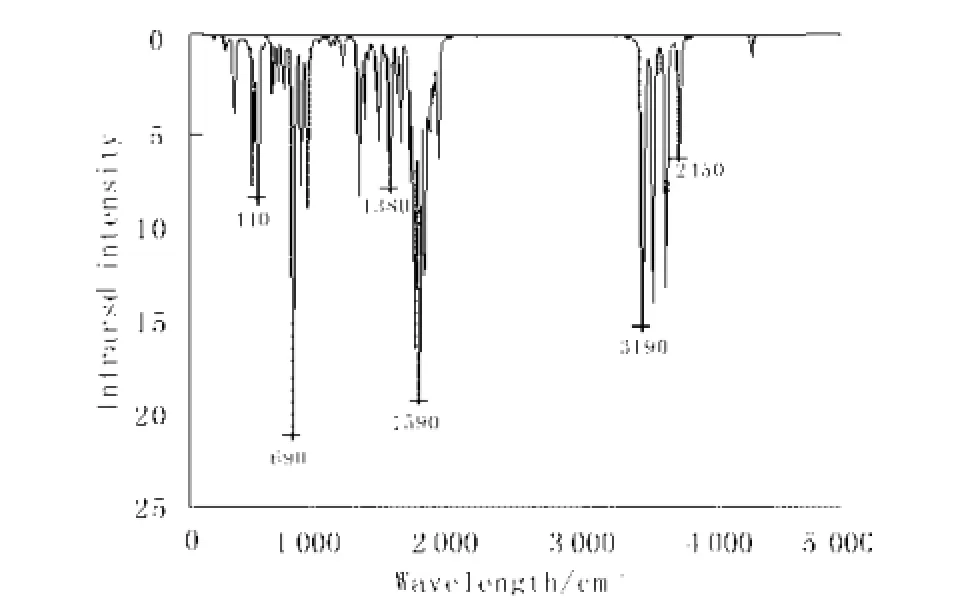
图 3 模拟甲基苯丙胺分子的IR图谱Fig.3 Simulation IR spectrum of methamphetamine

图4 模拟甲基苯丙胺分子的拉曼图谱Fig.4 Simulation Raman spectra of methamphetamine
2.4 核磁共振光谱
理论模拟了甲基苯丙胺分子的氢核磁共振谱(见图5所示),其苯环上五个氢(弱,7.12×10-6),主链CH2(中,2.52,2.76×10-6); 支链CH3(中,1.10× 10-6); 胺基上的CH3(强,2.74×10-6);胺基氢(中,2.0 ×10-6) 。这些峰较好地与其实验值吻合起来[2],也进一步验证了计算的准确性。

图5 模拟甲基苯丙胺分子的核磁共振谱Fig.5 Simulation 1HNMR of methamphetamine
2.5 荧光发射光谱
用TD DFT/B3LYP 6-31+G//CIS HF6-31+g方法,对标题物分子进行了分子荧光模拟, 发现化合物在235.0 nm处的紫外光激励下,能产生出弱的荧光发射强度,理论峰值位于331.5 nm(图 6,实验值
为360 nm)[4], 量子化学模拟计算发现,该分子的发光属于苯环内电子之间的π-π* 跃迁所为。
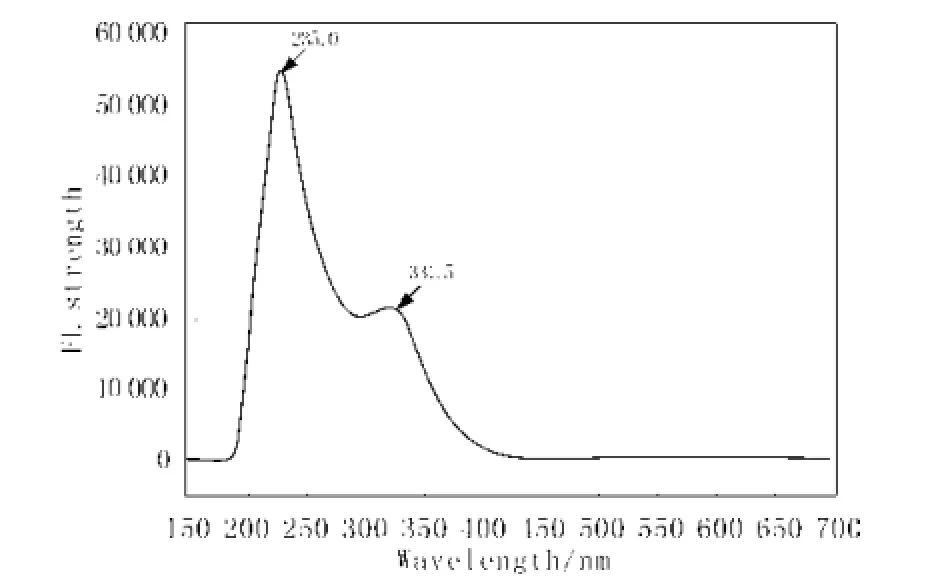
图6 模拟标题物分子荧光Fig. 6 Simulation FL of methamphetamine
2.6 反应活性
原子的自然电荷是研究分子的化学活性点位及确定活性区域的亲核或亲电特性强弱的有效方法。表 1的数据显示,胺基氮(N9)电荷最负(-0.320e),其次为与胺基氮相连的碳(C8,-0.171e);电荷最正的是胺基上的氢原子(H19, 0.12e)。自然电荷计算表明,胺基(NH)原子很可能是其发挥药理和药理活性的亲电和亲核反应中心。
3 结 论
对甲基苯丙胺分子采用密度泛函理论和方法,在6-311+G(d,p)基组水平上进行了优化计算。对其分子光谱(UV-Vis,IR, 1HNMR 以及荧光光谱)进行了理论模拟和指认,取得了与实验基本吻合的结果。自然电荷计算表明,胺基(-NH)氮和氢原子很可能是其发挥药理活性的亲电和亲核反应中心。以上理论模拟光谱峰及其指认将为甲基苯丙胺分析与检测提供帮助。

表1 甲基苯丙胺分子的自然电荷Table 1 Molecular natural charges of methamphetamine
[1]赵金涛, 陈大鹏, 张鹏翔,等. “冰毒”拉曼散射振动模式的研究[J].光谱学与 光谱分析,1999,15(5): 687-690.
[2]姚红艳,阙玉和,王思宏. 盐酸甲基安非他命的分析[J].化学世界,1999, 18:568-570.
[3]张金庄. 基于红外光谱无损检测自封袋表面微量毒品的研究[J]. 中国人民公安大学学报(自然科学版), 2013,75(1):14-16.
[4]郑天. 荧光分光光度法测定缴获冰毒的纯度[J]. 江苏警官学院学报, 2013,28(2):103-105.
[5]张润生, 王跨陡, 龚飞君,等. 苯丙胺类毒品及其衍生物的气相色谱-红外光谱分析[J]. 分析化学,2012, 40(6):915-919.
[6]傅强,廖林川,陈礼莉. HPLC法测定甲基苯丙胺与苯丙胺[J]. 四川大学学报(医学版), 2007,38(6):l025-1028.
[7]王继芬, 余静, 孙兴龙, 等. 毒品及其常见添加成分的拉曼光谱快速分析[J]. 光散射学报, 2012, 24(3):312-315.
Density Functional Analysis and Spectral Identification of Methamphetamine
CHEN Kai-hao,ZHONG Ai-guo
(College of Pharmaceutical and Chemical Engineering, Taizhou University , Zhejiang Taizhou 316000,China)
Using density functional theory DFT/B3LYP/6-311 + G (d, p) method and basis set, UV-Vis spectroscopy, IR spectroscopy,1HNMR spectrum and fluorescence spectroscopy of methamphetamine were studied by the theoretical simulation and identification. Natural charge calculation shows that N and H atoms of amine may be electrophilic and nucleophilic reaction center to play the pharmacological activity.
Ice; Density functional theory; Electronic spectra
O 641
: A
: 1671-0460(2014)01-0029-03
2013-11-18
陈凯浩(1990-),浙江奉化人,台州学院化学教育专业,研究方向:计算化学。E-mail:zhongaiguo@tzc.edu.cn。
钟爱国(1964-),男,研究方向: 计算化学。E-mail:xg2268@163.com。
