γ-氨基丁酸受体同源模建及与黄酮类化合物的分子对接
2014-02-27巨修练
巨修练,钱 程
1.武汉工程大学化工与制药学院,湖北 武汉 430074;2.绿色化工过程教育部重点实验室(武汉工程大学),湖北 武汉 430074
黄酮类化合物(如图1)具有突出的抗焦虑效果,且不引起肌肉松弛、健忘等副作用.这些重大发现推动了对选择抗焦虑,亲和力高的配体研究.6-溴黄酮相对于黄酮对GABAA受体的苯二氮唑结合位点的亲和力增加了近10倍[1].6-溴黄酮和GABAA的竞争结合参数是1.6~2.0(通过加入或不加入GABA时测量而得的6-溴黄酮和苯二氮唑结合位点的作用值比),与地西泮很类似,具有完全激动剂的药理学特征[2].当引入硝基时甚至会得到更有效的配体,6,3’ -二硝基黄酮与苯二氮唑位点结合力更强,其Ki值取值范围为17~50 nmol/L,它具有极有效的抗焦虑和肌肉松弛作用.6-溴-3’-硝基黄酮相对于双硝基类似物,对GABAA受体苯二氮唑结合位点的亲和力稍有增加,对苯二氮唑位点同样具有选择性[3].这类化合物的抗焦虑作用尽管比6,3’-二硝基黄酮稍差,但仍比地西泮的抗焦虑作用强.由于黄酮类化合物药理学选择性及其自身对GABAA受体苯二氮唑结合位点的活性低,使得对其衍生物的研究有了一个很大的飞跃,同时也对GABAA受体苯二氮唑结合位点选择性有了新的认识,促进了黄酮类化合物的发展.
本研究采用海兔乙酰胆碱结合蛋白(2XYS)作为模板,对人α1β2γ2 GABAA受体进行模建并能量优化,利用分子对接进一步指导研究.此外,从对接结果进行分析,发现黄酮类化合物的3’位对活性影响很大,且增多取代基团也会提高活性,进而为更好的设计黄酮类化合物提供了理论依据.
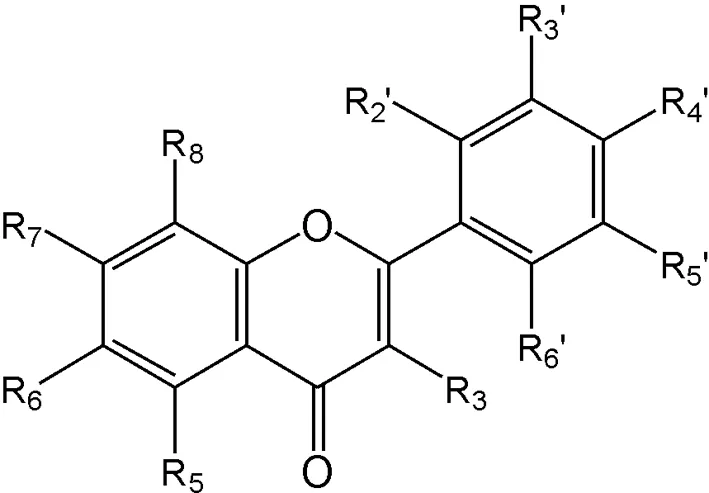
图1 黄酮类化合物的基本结构Fig.1 Structure of flavonoid
1 实验部分
本研究的所有计算实验都是通过SYBYL-X1.2软件(Tripos Inc.)完成,若无特别介绍,参数均采用缺省值.
1.1 人α1β2γ2GABAA受体同源模建
1.1.1 同源模建简介 大多数受体蛋白的三级结构尚未解析,并且同源蛋白在进化过程中保持着结构保守性,利用同源蛋白作为模板来构建受体蛋白是一种有效的方法[4].常用的软件有两类:一是利用蛋白质分析软件来预测,例如SWISS-MODEL主要用于预测蛋白质的高级结构,它通过同源建模的方法可对未知序列的三级结构进行预测;另一类是对蛋白的结晶进行X-衍射、核磁共振技术及冷冻电镜等方法获得结构信息,将已知的蛋白质立体结构为模板,利用相关软件构建已知序列的蛋白高级结构,例如SYBYL, Discovery Studio,Modeller都是比较常用的软件 .
1.1.2 同源模建a.序列选择 为了模建目标受体人α1β2γ2GABAA受体,首先通过Swiss-Prot/TrEMBL数据库筛选出合适的亚基序列:人α1亚基(蛋白质序列号:P14867),β2亚基(蛋白质序列号:P47870),γ2亚基 (蛋白质序列号:P18507),然后对以上3种亚基进行编辑.考虑到所构建的受体在跨膜区,所以删去膜内环区与膜外结合区域的氨基酸序列.
b.模板选择 模板在同源模建中起关键作用,它直接影响靶结构的质量,并对结果起决定性作用[5].由于膜介蛋白晶体结构较难获得,且当前结构解析技术直接决定了膜介蛋白结构的分辨率,因此已解析的三维结构膜蛋白数目十分有限.本实验中,采用海兔乙酰胆碱结合蛋白作为模板(PDB登记号2XYS,0.194 nm)[6].
c.构建亚基 首先采用SYBYL-X1.2中的Biopolymer模块中的compare sequences将靶标序列与模板序列进行比对(sequence alignment),从而生成MSF (multiple sequence format)文件[7].然后再将Model Proteins中的ORCHESTRAR模块中导入MSF文件,将模板结构与靶序列的三维结构进行结构比对.最后构建靶肽链的结构,包括搜索环区,识别结构保守区域(structure conserved regions,SCR)和添加侧链.
d.模型组合 将人α1β2γ2受体与模板2XYS进行比对,需要将前者的α1亚基β2亚基γ2亚基分别叠合到2XYS为模板的各个亚基上,以顺时针方向(β2)(γ2)(α1)(β2)(γ2)构建人α1β2γ2GABAA受体,如图2所示.

图2 人α1β2γ2 GABAA亚基对应示意图Fig.2 Subunit of human α1β2γ2 GABAA
e.模型的优化与修正 因为只有运用分子动力学与分子力学的方法对受体进行修正,才能验证受体的可靠性.因此,本实验选取立体场AMBER7 FF99[8],运用共轭梯度法优化体系能量梯度的RMS小于5 kcal/mol/nm,并通过分子动力学(molecular dynamic, MD)优化模型,以检验模型的稳定性.计算条件设置为300 K恒温,每2.5 ps采样一次轨迹数据,步长1 fs,计算总长为500 ps.
1.2 分子对接
1.2.1 对接方法 本实验首先利用分子对接法将前面讨论的黄酮类配体化合物与人α1β2γ2 GABAA受体蛋白对接.并结合打分函数,研究配体化合物与受体蛋白之间的相互作用,探究结合自由能与实测生物活性的线性关系,同时验证模型的可靠性.
Surflex-docking[9]是SYBYL-X1.2中的一个模块,可用于计算配体与受体之间的相互作用能,也可用于复合物的结构优化.本研究利用Surflex-docking进行分子对接实验. 因为受体的结合口袋直接影响分子对接结果,所以定义结合口袋具有十分重要的意义[10].前文中同源模建人的α1β2γ2GABAA亚型,选用共晶的6-氟-3’-硝基黄酮作为活性区域,利用残基模式定义活性结合位点. Surflex-docking模块中,原型分子(protomol)即我们所称的活性位点.此外,bloat与threshold这两个参数直接影响原型分子的大小与形状. 其中,Bloat影响原型分子渗入蛋白空隙的程度,Threshold影响原型分子的大小[11].
1.2.2 对接配体分子 通过SYBYL-X1.2软件中的dock ligand模块,41种黄酮类化合物(如图7)对接到模建的α1β2γ2GABAA受体中.
Surflex-docking进行对接实验后,分子配体的对接结果可通过Csorce模块显示.打分函数基于受体-配体复合物的结合亲和力,将会考虑熵作用,极性作用,疏水作用,排斥作用,溶剂化作用.最后给出的总分即-lg10Kd,Kd为配体的解离常数.可通过以下函数计算配体与受体的结合自由能(Free Energy of Binding, kcal/mol):
Free Energy of Binding=RTlnK[12-13]
2 结果和讨论
2.1 同源模建
将海兔乙酰胆碱结合蛋白与α1β2γ2进行序列比对,得到其同源性(Identity)分别为19.3%,19.6%和16.0%,由此可知:选取的2XYS作为人α1β2γ2亚基模型是比较合理的.将人α1β2γ2 GABAA受体与海兔乙酰胆碱结合蛋白进行序列比对,如图3所示.

图3 人α1β2γ2GABAA受体与海兔乙酰胆碱结合蛋白序列比对图Fig.3 Result of sequence alignment between human α1β2γ2GABAA and aplysia californica nAChR
经过比对分子序列,搜索保守区域、构建疏水环区、添加分子侧链、分子动力学优化后,得到人α1β2γ2 GABAA的二级结构,如图4所示.

图4 人α1β2γ2GABAA受体二级示意图Fig.4 Secondary structure of human α1β2γ2GABAA
人α1β2γ2GABAA受体需要进行结构修正和能量优化.通过ProTable验证受体模型的立体化学性质.人α1β2γ2GABAA的受体模型拉氏构象图如图5所示.
从以上拉氏图可知,大量的氨基酸残基聚集在值为-150度与-40度处,人α1β2γ2 GABA A受体模型中有96.76%的氨基酸残基处于允许区域,验证了模型的合理性.因此,实验中模建的人α1β2γ2 GABAA受体模型是可靠的,可用作分子对接实验.
利用SYBYL-X1.2的Dynamics模块进行分子动力学模拟,人α1β2γ2 GABA A受体模型的动力学模拟能量-时间图如图6.经过分子动力学能量优化后,能量-时间图里可知,α1β2γ2 GABA A受体模型在前300 ps能量变化较大,而后200 ps能量比较稳定,通过分子动力学实验,也说明了该模型是可靠的[14-15].
2.2 分子对接
本实验利用分子对接法将黄酮类化合物与人α1β2γ2 GABAA受体进行对接.通过上文软件打分函数评价对接结果.
利用SYBY-X1.2软件包中Surflex-docking模块进行分子对接以及氢键,通过分子对接和打分函数的方法对打分函数进行评价,得到黄酮类化合物与人α1β2γ2 GABAA受体的对接得分,如表1所示.

表1 黄酮类化合物的对接得分Tabel 1 Docking result of Flavonoids
由于13号分子打分较高,且结构相似性较高具有代表性,故对其与人α1β2γ2 GABAA受体对接图(图7),进行分析.

图7 人α1β2γ2 GABAA受体与6-氟-3’-硝基黄酮对接图Fig.7 Human α1β2γ2 GABAA docking with 6-fluoro-3’-nitro flavonoid
从上图可知,13号分子3’位硝基的一个氧原子和氮原子作为氢键受体,与Arg97(精氨酸)侧链上的亚胺上的氢原子形成氢键,为配体分子与受体结合提供了条件,同时也验证了3’位对黄酮类化合物活性影响的重要性.而且从空间构象来看,13号分子的母环与Tyr190(酪氨酸)和Tyr140(酪氨酸)的苯环形成共轭,这样能使体系的能量降低,分子稳定,也说明了所建模型的合理性.同时,从表1中可以发现,对比1号分子与9号分子,2号分子与10号分子,3号分子与11号分子,7号分子与13号分子可发现,当6位所连基团相同时,将3’位的氢原子替换成硝基时,对接打分明显提高.同理可看出,将3’位替换为其他大分子基团时,对接打分也有所提高.且大部分化合物的对接打分与实测活性一致,也验证了模型的可靠性.综上所述,黄酮类化合物3’位对活性影响很重要.同时,38号化合物的高得分,可能与较多的取代基提高了与结合口袋的结合能力有关.
3 结 语
本研究通过同源模建人α1β2γ2 GABAA受体跨膜区的三维结构,并结合分子动力学优化和能量优化,验证了所建模型的稳定性与可靠性.进而将41个黄酮类衍生物与该模型进行分子对接实验,通过对接结果的分析,得知当母环3’位被取代时,分子活性有所增加,其中被硝基取代时,活性提高较大.并从38号化合物中得到提示,相应的增加取代基的个数,可能会提高活性,为设计黄酮类化合物提供了理论依据.
致 谢
本实验基于武汉工程大学化工与制药学院提供的计算平台,在此表示感谢!
[1] ARGYROPOULOS S V, NUTT D. The use of benzodiazepines in,anxiety and other disorders [J]. The journal of the European College of Neuropsychopharmacology, 1999, 9 (6): 407-412.
[2] VIOLA H, MARDER M, WOLFMAN C, et al. 6-Bromo-3’-nitroflavone, a new high affinity benzodiazepine receptor agonist recognizes two populations ofcentral cortical binding sites [J]. Bioorganic and Medicinal Chemistry Letters, 1997, 7 (3): 373-378.
[3] CRISTINA W, MARIEL M. Flavonoids as GABAA receptor ligands:the whole story[J]. Journal of Experimental Pharmacology, 2012, 4: 9-24.
[4] MARSHALL G R, MAYER D, NAYLOR C B, et al. Mechanism-based analysis of enzyme inhibitors of amide bond hydrolysis [J]. Progress in Clinical and biological research, 1989, 291: 287-295.
[5] ZHU Z Y, SALI A, BLUNDELL T L. A variable gap penalty function and feature weights for protein 3-D structure comparisons [J]. Protein Engineering, 1992, 5 (1): 43-51.
[6] BRAMS M, PANDYA A, KUZMIN D, et al. A structural and mutagenic blueprint for molecular recognition of strychnine and d-tubocurarine by different cys-loop receptors [J]. PloS Biology, 2011, 9 (3): 1100-1034.
[7] NEEDLEMAN S B,WUNSCH C D. A general method applicable to the search for similarities in the amino acid sequence of two proteins [J]. Journal of Molecular Biology, 1970, 48 (3): 443-453.
[8] WANG J M, WOLF R M, CALDWELL J W, et al. Development and testing of a general amber force field [J]. Journal of Computational Chemistry, 2004, 25 (9): 1157-1174.
[9] RUPPERT J, WELCH W, JAIN AN. Automatic identification and representation of protein binding sites for molecular docking [J]. Protein Science, 1997, 6 (3): 524-533.
[10] 巨修练,王黎丽,李科.斑马鱼A型γ-氨基丁酸受体同源模建及分子对接[J].武汉工程大学学报,2013(6):20-29.
JU Xiu-lian, WANG Li-li, LI Ke. Homology modeling and docking of zebrafish γ-aminobutyric acid receptor [J]. Journal of Wuhan Institute of Technology, 2013 (6):20-29.(in Chinese)
[11] CRAMER R D, PATTERSON D E, BUNCE J D. Recent advances in comparative molecular field analysis (CoMFA) [J]. Progress in Clinical and Biological, 1989, 291: 161-165.
[12] CORRINGER P J, BAADEN M, BOCQUET N, et al. Atomic structure and dynamics of pentameric ligand-gated ion channels: new insight from bacterial homologues [J]. The Journal of Physiology, 2001, 588 (4): 565-572
[13] CHEN L G, DURKIN K A, CASIDA J E. Structure model for gamma-aminobutyric acid receptor noncompetitive antagonist binding: widely diverse structure fit the same site [J]. Proceedings of the National Academy of Science of the United Stated of America, 2006, 103 (13): 5185-5190.
[14] MITEVA M A, LEE W H, MONTES M O, et al. Fast structure-based virtual ligand screening combining FRED, DOCK, and Surflex [J]. Journal of Medicinal Chemistry, 2005, 48 (19): 6012-6022.
[15] CHENG Jin, JU Xiu-lian, CHEN Xiang-yang, et al. Homology modeling of human α1β2γ2 and house fly β3 GABA receptor channels and Surflex-docking of fipronil [J]. Journal of Molecular Modeling, 2009, 15: 1145-1153.
