脊髓性肌萎缩的诊疗进展
2014-02-14刘倩综述吴菁审校
刘倩 综述 吴菁 审校
(广东省妇幼保健院医学遗传中心,广东广州 510010)
脊髓性肌萎缩的诊疗进展
刘倩 综述 吴菁 审校
(广东省妇幼保健院医学遗传中心,广东广州 510010)
脊髓性肌萎缩(spinal muscular atrophy,SMA)是一种基因突变导致脊髓前角细胞变性引起肌无力和肌萎缩等临床症状的一组疾病。遗传方式以常染色体隐性遗传为主,也有常染色体显性遗传和X连锁遗传[1,2]的报道。活产儿中发病率约为1/6000~1/10 000,是第二常见的致死性常染色体隐性遗传病,仅次于囊性纤维化。文献显示,中国南方SMA致病基因SMN1携带者率为1/35~1/80[3],与国外学者[4]报道的携带者发生率1/40~1/60相似。SMA的诊断过程复杂,缺乏有效的治疗办法,防治SMA的有效途径是进行产前诊断,避免患儿的出生,或通过辅助生育技术进行植入前诊断。现就该病的诊断与治疗进展进行综述。
1 临床表现与分型
SMA临床上主要表现为肌肉萎缩和肌张力减低进行性加重。根据疾病严重程度及发病年龄,1992年欧洲神经肌肉疾病中心召开的SMA国际研讨会将其分为4型。
1.1 Ⅰ型 婴儿型,又称Wardning-Hoffmann病,多于出生后6个月内发病,起病急、病程进展快,表现为严重的全身肌无力和肌张力不全,不能独坐或行走,常于2岁内死于呼吸麻痹或肺部感染等。
1.2 Ⅱ型 中间型,通常在出生后6~18个月起病,患儿可独坐,但不能独自站立或行走,多数可存活至2岁以后。
1.3 Ⅲ型 少年型,又称Kugelberg-Welander病,常于2~17岁起病,多数仅表现为轻度肌力减弱,可独自站立和行走。起病后逐渐失去行走能力,病程进展缓慢,可存活至成年。
1.4 Ⅳ型 成年型,一般于30岁以后发病,表现为缓慢逐渐发生的上下肢近端无力和肌萎缩,有肌震颤,以骨盆带肌和肩胛带肌为著,出现上楼困难以及梳头无力等。40岁以后才发病者可累及肢体远端。
2 分子遗传学和病因
1990年Brzutowicz等[5]将SMA的致病基因定位于5q11.2~q13.3。1995年Lefebvre等[6]学者在该基因座区域检测到4个不同的c DNA克隆,证实运动神经元存活基因(survival motor neuron,SMN)是致病基因。SMN基因全长约27 kb,含9个外显子(exon 1、2a、2b、3-8),有2个高度同源拷贝SMN1和SMN2,端粒侧称SMNT(telomeric SMN)或SMN1,着丝粒侧称SMNC(centromeric SMN)或SMN2。两者仅在各自的3′端有5个碱基的差别,其中2个碱基位于外显子7、8,另3个碱基在内含子6、7[7]。神经元凋亡抑制基因(NAIP)基因编码神经元凋亡抑制蛋白,是SMA的修饰基因。
SMN1基因的缺失是SMA的基础发病机制,SMN2基因和NAIP基因的异常则与疾病严重程度相关[8]。约95%SMA患者存在SMN1基因第7号外显子的纯合性缺失[9,10]。疾病严重程度与SMN2拷贝数相关,大部分Ⅰ型患者有2个拷贝数,Ⅱ型患者有3个拷贝数,Ⅲ型和Ⅳ型有3到4个拷贝数。NAIP基因的第5、6号外显子可能与SMA的临床表型有关。
SMN1编码全长的SMN转录产物(SMN-fl),而SMN2编码生成大量跳跃外显子7的选择性转录产物(SMN△7)和少量SMN-fl。SMN-fl编码一个包涵294个氨基酸,分子量为38k D的蛋白,称为SMN蛋白。SMN蛋白广泛分布于全身组织中,表达水平各异,其中脑、脊髓和肌肉中的表达水平最高。SMN基因缺失或突变时,其表达产物SMN蛋白数目、结构、功能均异常。SMN蛋白数量低于运动神经元所需最低含量时,将发生运动神经元变性。因此,推测SMA的机制可能为SMN基因缺失或突变导致SMN蛋白数目、结构或功能异常,引起运动神经元变性,从而导致肌无力、肌萎缩。
3 诊断
3.1 临床表现 SMA的主要诊断依据是临床表现,特别是症状严重的患儿。患儿一般无智力低下,肌力的下降通常为对称性的,近端重于远端,下肢重于上肢,疾病的严重程度与发病年龄相关。临床上是否易被发现以及腱反射的消失与疾病的年龄背景相关。严重病例的临床特点包括:哭声或咳嗽无力,喂养困难,不能抬头等。临床上接诊到与SMA临床特点一致或相似的病人时,诊疗流程如图1。
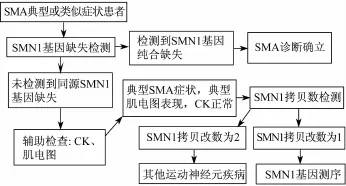
图1 脊髓性肌萎缩的诊断流程图
3.2 基因检测 对怀疑SMA的患者进行的第一层诊断性检测为SMN1基因纯合缺失的检测。SMN1基因外显子7缺失(伴或不伴外显子8的缺失)可被诊断为SMA,敏感性95%,特异性100%[11]。在SMA患者中最常见的突变是SMN1基因的纯合缺失,大部分携带者可检测到SMN1基因其中一个等位基因的缺失,对SMN1基因外显子7、8进行检测,是SMA基因诊断和产前诊断的首选方法。基因缺失的检测方法包括单肽链构象多态性PCR(PCR-SSCP)、错配PCR-限制性片段多态性(PCR-RFLP)、等位基因特异性PCR(AS-PCR)、变性高效液相色谱(DHPLC)、多重依赖性探针扩增法(MPLA)等。分子细胞学的检测敏感性为93%~95%,部分潜在SMA患者在出现典型临床症状前可能无法检出。
在众多SMA的分子诊断技术中,PCR-RFLP最常用,也是最初用于确定SMA治病基因的基因检测方法。可根据SMN1和SMN2基因外显子间碱基差别设计错配引物,并根据特异的酶切位点判定SMN1基因是否存在纯合缺失。但该方法有一定局限性,无法检出携带者的杂合缺失[12]。
MLPA是2002年荷兰学者Schouten等发明的一项基因半定量分析技术,可对致病基因及修饰基因进行定量分析。陈雅芳等[13]通过比较MLPA与PCR-RFLP两种基因检测方法,得出结论:MLPA是简便、高校、可靠的基因定量分析技术,在SMA的诊断、携带者筛查及产前诊断工作中是一种有效的基因检测方法。
DHPLC是一种高通量、自动化的基因检测技术,已有国内外学者运用DHPLC与PCR-RFLP两种方法同时检测SMA患儿及正常对照血清,证实DHLPC省时、高灵敏、特异性强的检测方法。可以准确诊断出SMN基因的拷贝数,可作为患者及携带者筛查的检测方法[14,15]。
3.3 辅助检查 当基因检测结果阴性时,进一步进行实验室相关检查,包括肌酸激酶(creatine kinases,CK)、肌电图(electromyography,EMG)和神经传导检测。如果EMG检测提示为运动神经元疾病,则进一步进行SMN突变检测。通过多重连接酶探针依赖扩增(multiplex ligation-dependent probe amplification,MLPA)或实时定量PCR方法可检测SMN1基因拷贝数,这种半定量检测方法可将敏感性提高到98%[16]。如果患者仅有1个SMN1拷贝,需对无缺失的等位基因进行测序用以确定是否含有较少见的病因(突变),常见的基因微小变异包括:点突变、插入和缺失。然而,约有1/3的患者有典型的临床表现和SMN1 1个拷贝,在SMN1/SMN2编码区未发现其他突变。这一特征在SMAⅢ型患者中更常见,也许和更深层次的基因内突变有关,目前尚未证实。
4 产前诊断
SMA是常见遗传性疾病之一,携带者频率约为1/50,缺乏有效的治疗手段,携带者基因筛查并进行必要的产前诊断是公认的预防措施。对高危人群进行详尽的遗传咨询并进行产前诊断,是降低SMA出生率的有效方法。Mahmoud等[17]指出对于SMN1基因杂合缺失突变携带的夫妻,SMN1基因缺失的分析产前诊断准确率接近100%。曾经生育过SMA患儿的夫妇,SMA患儿的再发风险为25%,在以后的每次妊娠期间均需进行产前诊断。可通过绒毛活检术、羊膜腔穿刺术或脐静脉穿刺术对胎儿进行取样,并对胎儿样品进行SMN基因检测,对于确诊的SMA胎儿,可通过知情同意择时实施终止妊娠,避免其出生,以达到防治出生缺陷,提高人口素质的目的。
刘新秀等[18]对17~20周有SMA生育史的孕妇进行羊膜腔穿刺术,应用PCR-RFLP方法检测SMN1基因缺失情况,并用短串联重复序列(short tandem repeats,STR)进行母血污染鉴定。黄欢等[19]自行研制“Hb H-Buffer”,对羊水直接扩增,结果显示,与提取羊水DNA后的扩增效率一致。扩增后通过PCR-RFLP方法进行分析SMN基因缺失情况。应用羊水细胞直接扩增法,可减少样本量,简化步骤,减少污染可能,并可实现快速产前诊断。在SMA的产前诊断中是一种值得推广的技术。
植入前诊断(Preimplantation genetic diagnosis,PGD)是预防遗传性疾病并可避免终止妊娠术的一种产前诊断方法。但用于PGD的单个二倍体细胞所能获取到的DNA量有限,导致了包括检出污染率增高、等位基因扩增失败等在内的一系列问题。STR具有可扩增、高度多态性等特点,克服了这些限制。在此,文献报道适于SMA检测的侧翼基因区的五个STR杂合性分析标记(D5S1408,D5S1417,D5S610,D5S637)。这些STR位点符合Hardy温伯格平衡。可将类似方法用于其他单基因病的植入前诊断[20]。
2008年美国医学遗传学会(American College of Medical Genetics,ACMG)的指南[21]指出:目前,仅有SMA家族史阳性的群体接受了相关基因携带的筛查,推荐应用到更广范围的人群筛查。因其满足进行人群筛查的五大关键因素,即临床表现严重、人群携带率高、有灵敏特异的可靠检测方法、可进行产前诊断和遗传咨询等。国内,有产前诊断资质,有条件开展SMA致病基因筛查及诊断的单位,如能进行人群SMN基因筛查,针对双方均为携带者的夫妇进行产前诊断,避免SMA患儿的出生,能更有效降低SMA患儿出生率。从而达到降低出生缺陷、提高人口素质的目的。
5 治疗
由于SMA的病理生理学机制未明,目前暂无有效治疗办法。在过去的数十年间,SMA的治疗以支持性护理措施为主,无有效治疗方法。近年来,随着SMA相关分子学机制研究的不断深入,几种治疗方法在不断发展完善,治疗策略包括依赖SMN的治疗和不依赖SMN的治疗[22]。
5.1 依赖SMN的治疗 依赖SMN的治疗策略是通过药物、RNA、反式剪接RNA、干细胞等,改善SMN2基因表达以及SMN蛋白含量,从而达到改善SMA症状的目的。
5.1.1 药物治疗 一种潜在的SMA治疗模式是利用小分子物质,增加外显子7的插入、激活SMN2启动子、延长SMN mRNA或SMN蛋白半衰期、延长SMN蛋白的C端,或整合以上分子活动。新型喹唑啉衍生物及组蛋白去乙酰化酶(HDAC)抑制剂是最新开发的新药,前者尚在动物实验阶段[23],后者处于临床研究阶段[24]。
5.1.2 基于RNA的治疗 利用小分子RNA对异常m RNA进行间接重排,这是包括SMA在内的常见遗传性疾病的扩展研究领域的焦点。调节SMN2m RNA可恢复适当的SMN蛋白的表达,从而达到改善SMA患者症状的目的。
本领域的研究主要围绕反义寡核苷酸(ASOs)展开,根据SMN2基因的结构特点,利用ASOs拼接结合成双功能RNA基团[25-27],在小鼠模型侧脑室内注射这些双功能RNA基团,可使鼠脑内SMN蛋白含量增加[28]。由载体运载的双功能RNA基团到达神经系统后,小鼠脑和脊髓的SMN蛋白含量可与杂合突变基因携带者SMN蛋白含量相媲美[29]。
5.1.3 反式剪接RNA SMN反式剪接是RNA治疗外的一种很有前景的治疗方法。反式-拼接需要组成的3个域的合成RNA(tsRNA):①域名绑定进行交互与特定的目标;②拼接的域进行剪接反应与所选的内含子;③完整外显子或一系列的外显子以更换有缺陷的基因片段。反式-拼接的产品是嵌合mRNA,转化为有功能的蛋白质。向严重的SMA小鼠体内注射tsRNA后明显改善疾病严重程度并延长寿命70%[30]。
5.1.4 干细胞 近年来备受关注的SMA治疗策略是用干细胞进行细胞替代治疗。细胞替代是通过移植经过体外或内源性干细胞激活的中枢神经系统干细胞来实现的。目前干细胞治疗仅有骨髓移植和间充质细胞移植,尚无应用于SMA的研究报道。脊髓神经干细胞的研究已取得长足进步,有学者发现脊髓神经干细胞可改善SMA鼠模型的表型,但仅限于基础研究,尚无法推广到人[31]。在另一项研究中,对较严重的SMA鼠模型注射胚胎干细胞来源的神经细胞前体,源于多能干细胞的胚胎干细胞表现出治疗潜能[32]。近来,从病人的成纤维细胞中成功诱导出多能干细胞(iPS),是研究用于干细胞治疗的基因兼容神经元的一个重大进步。
5.2 不依赖SMN的治疗
5.2.2 肌肉增强 肌肉增强被认为是SMA治疗的目标。有学者研究发现,人血清白蛋白基因启动子可控制转基因动物肌肉中SMN的表达,肌肉中表达的SMN独立于骨骼肌之外,与SMA分型不冲突。可能机制是增强的肌肉有助于内在运动单位的维持和稳定[33]。两个研究比较了各种肌肉生长抑制剂的抑制素通路,其中一个研究证明重组卵泡休止素释放后,寿命和运动功能可有中等程度改善;另一个研究,检测到Act RIIB-Fc或重组卵泡休止素治疗后的SMNΔ7小鼠,表型未改变[34,35]。存在这种差异的基础尚不清楚,可能是运动神经元需要额外的支持,以最佳方式响应基于SMN的治疗。
5.2.3 肌动蛋白动力学 有学者使用一种称为Smn2B的新型温和SMA鼠模型进行研究,经Rho激酶抑制剂治疗后,生存时间急剧增加,神经肌肉接头也被增强,并在后来的治疗中更成熟。值得注意的是,这些表型的改变与SMN的增加无关。这些研究结果不仅提供新的治疗靶点,还可能丰富SMN功能的视野。
6 展望
脊髓性肌萎缩是一种常见的常染色体隐性遗传性疾病,目前暂无有效治疗方法,严重类型起病急危害重,做好疾病的诊断与产前诊断,尽量避免更多患者的出现是预防SMA的有效方法。如能对人群展开筛查,对携带者夫妻进行遗传咨询及产前诊断,则更能有效地降低脊髓性肌萎缩的发病率。研究出有效治疗方法,也是SMA研究的一个重要方向。
[1]Jiang W,Ji X,Xu Y,et al.Molecular prenatal diagnosis of autosomal recessive spinal muscular atrophies using quantification polymerase chain reaction[J].Genet Test Mol Biomarkers 2013,17(5):438-442.
[2]Baumbach RL,Sacharow S,Ahearn ME.Spinal Muscular Atrophy,X-Linked Infantile[M].GeneReview.Scattle:University of Washington,Seattle,1993-2014.
[3]Chan V,Yip B,Yam I,et al.Carrier Incidence for Spinal Muscular Atrophy in Southern Chinese[J].Journal of Neuronlogy,2004,251(9):1089-1093.
[4]Prior TW,Snyder PJ,Rink BD,et al.Newborn and carrier screening for spinal muscular atrophy[J].Am J Med Genet A,2010,152A:1605-1607.
[5]Brzustowicz LM,Lehner T,Castilla LH,et al.Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3[J].Nature,1990,344:540-541.
[6]Lefebvre S,Burglen L,Reboullet S,et al.Identification and characterization of a spinal muscular atrophy-determining gene[J].Cell,1995,80:155-165.
[7]Burglen L,Lefebvre S,Clermont O,et al.Structure and Organization of the Human Survival Motor Neurone(SMN)Gene[J].Genomics,1996,32:479-482.
[8]Burglen L,Lefebvre S,Clermont O,et al.Structure and Organization of the Human Survival Motor Neurone(SMN)Gene[J].Genomics,1996,32:479-482.
[9]Lefebvre S,Burglen L,Reboullet S,et al.Identification and characterization of a spinal muscular atrophy-determining gene[J].Cell,1995,80(1):155-165.
[10]Vitte J,Fassier C,Tiziano FD,et al.Refined characterization of the expression and stability of the SMN gene products[J].Am J Pathol,2007,171:1269-1280.
[11]Wang CH,Finkel RS,Bertini ES,et al.Participants of the International Conference on SMA Standard of Care.Consensus statement for standard of care in spinal muscular atrophy[J].J Child Neurol,2007,22:1027-1049.
[12]张菁菁,罗春玉,刘安,等.应用MLPA技术进行脊髓性肌萎缩症高风险胎儿的产前诊断[J].现代妇产科进展,2014(1):47-49.
[13]陈雅芳,何瑾,张奇杰,等.应用多重连接依赖性探针技术进行脊髓性肌萎缩症产前诊断[J].中国现代神经病杂志.2012,2(3):294-299.
[14]李秀玲,贺静,朱宝生.脊肌萎缩症的基因诊断和产前诊断研究进展[J].中国妇幼保健,2011,26:2220-2223.
[15]肖雪,蔡兰云,王蕊艳,等.DHPLC技术在儿童型脊髓型肌萎缩症的基因诊断及携带者基因筛查中的应用[J].江西医学院学报,2009,49(2):99-102
[16]Arkblad EL,Darin N,Berg K,et al.Multiplex ligation-dependent probe amplification improbe diagnostics in spinal muscular atrophy[J].Neuromuscul Disord,2006,16:830-838.
[17]Mahmoud SK,Sima MD,Shamsei A.Prenatal diagnosis of spinal muscular atrophy:clinical experience and molecular genetics of SMN gene analysis in 36 cases[J].Journal of Prenatal Medicine,2013,7(3):32-34.
[18]刘新秀,陈万金,叶真,等,超声引导羊膜腔穿刺产前基因诊断脊髓性肌萎缩症[J/CD].中国产前诊断杂志(电子版),2011(01):13-16.
[19]黄欢,王珏,卢守莲,等,羊水直接PCR法快速诊断脊肌萎缩症的实验研究[J].现代生物医学进展,2013(35):6845-6847、6903.
[20]Korzebor A,Derakhshandeh-Peykar P,Meshkani M,et al.Heterozygosity assessment of five STR loci located at 5q13 region for preimplantation genetic diagnosis of spinal muscular atrophy[J].Mol Biol Rep,2013.40(1):67-72.
[21]ACMG Practice Guidelines.Carrier screening for spinal muscular atrophy[R].2008,10(11).
[22]Christian L,Hansjorg R,Monir S.Spinal muscular atrophy:mechanisms and therapeutic strategies[J].Hum.Mol.Genet.2010,19:111-118.
[23]Butchbach ME,Singh J,Thorsteinsdottir M,et al.Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy[J].Hum Mol Genet,2010,19:454-467.
[24]Swoboda KJ,Scott CB,Reyna SP,et al.Phase II open label study of valproic acid in spinal muscularatrophy[J].PLoS ONE,2009,4(5):e5268.
[25]Passini MA,Cheng SH.Prospects for the gene therapy of spinal muscular atrophy[J].Trends Mo Med.2011,17:259-65.
[26]Dominguez E,Marais T,Chatauret N,et al.Intravenous sc AAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice[J].Hum Mol Genet,2011,20:681-693.
[27]Passini MA,Bu J,Richards AM,et al.Antisense oligonucleotides delivered to the mouse CNSameliorate symptoms of severe spinal muscular atrophy[J].Sci Transl Med,2011,3:72ra18.
[28]Baughan TD,Dickson A,Osman EY,et al.Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy[J].Hum Mol Genet,2009,18:1600-1611.
[29]Dickson A,Osman E,Lorson CA.Negatively-acting bifunctional RNA increases survival motor neuron in vitro and in vivo[J].Hum Gene Ther,2008,19:1307-1315.
[30]Coady TH,Lorson CL.Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy[J].J Neurosci,2010,30:126-130.
[31]Corti S,Nizzardo M,Nardini M,et al.Embryonic stem cellderived neural stem cells improve spinal muscular atrophy phenotype in mice[J].Brain,2010,133:465-481.
[32]Corti S,Nizzardo M,Nardini M,et al.Motorneuron transplantation rescues the phenotype of SMARD1(spinal muscular atrophy with respiratory distress type 1)[J].J Neurosci,2009,29:11761-11771.
[33]Gavrilina,TO,McGovern VL,Workman E,et al.Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect[J].Hum Mol Genet,2008,17:1063-1075.
[34]Rose FF Jr,Mattis VB,Rindt H,et al.Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy[J].Hum Mol Genet,2009,18:997-1005.
[35]Sumner CJ,Wee CD,Warsing LC,et al.Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice[J].Hum Mol Genet,2009,18:3145-3152.
编辑:宋文颖
R714.55.1
A
10.13470/j.cnki.cjpd.2014.03.015
2014-05-26)
