Computational prediction of MHCⅡ-peptide ligands binding specificities by AUC Optimized Gibbs
2014-02-08SHENGHaoLUYufengZHANGYi
SHENG Hao, LU Yu-feng, ZHANG Yi
(1.School of Mathematical Sciences,Dalian University of Technology,Dalian 116024,China;2.School of Sciences,Hebei University of Science and Technology,Shijiazhuang 050018,China)
0 Introduction
Recently there has been constant concern about the rules for the binding of peptides to MHC molecules.The MHC molecules deliver fragmented pieces of an antigen protein on the host cell′s surface to the cytotoxic T cell(Tc)or the helper T cell(Th),giving rise to their development and activation.It is important to know which peptide fragments of pathogenderived proteins most probably bind to a certain MHC-molecule.The MHCⅠbinding groove is closed,which tends to bind short peptides of 8-10 amino acids by both ends.But the MHCⅡbinding groove is open,which makes the length of the peptides bound by MHCⅡmolecules unconstrained.And relative to the MHCⅠmolecules,the binding pockets of MHCⅡmolecules are more permissive in the accommodation of amino acids.The two obstacles greatly affect the performance of MHCⅡbinding peptide prediction.
On the opinion of the IBS hypothesis[1],for most peptides,each side chain of the peptide sequence contributes a certain amount to the stability of binding peptides to MHCⅡmolecules;and the MHCⅡ-peptide ligands binding affinity is independent of the peptide sequence.The influence of residues at each position in the peptide sequence on the binding affinity can be considered independently.Based on this hypothesis,some linear models,additive PLS method[2],stabilized matrix method[3],Gibbs sampling method[4]and SMM-align method[5]have achieved reasonable performance.
AUC Optimized Gibbs(AOG)method used in the study is a changed version of Gibbs sampling method.The Gibbs sampling method was applied to predict classⅠand classⅡepitopes[4];Whereas,the relative entropy is used to guide the sampler,resulting in that only binding peptides are used for training,and nonbinding peptides are discarded.This leads to a low efficient use of the training data.In the study,the homology reduced self-fitting AUC is used to guide the sampler,resulting in that both the positive and negative information could be incorporated into training the sampler.In the HLA-DR4(B1*0401)epitope benchmark and quantitative IEDB benchmark,the AOG algorithm is used as well as Gibbs sampling method[4]and TEPITOPE[6].Through reduction of the noise from the experimental data using the AOG method,MHCⅡbinding specificities are computationally predicted,and the profile of the MHCⅡmolecule interacting with its peptides is analyzed from the results of the algorithm.The processing of classⅡepitopes as well as design of better peptide vaccine can be understood well.
1 Methods and materials
1.1 Training and testing data
1.1.1 Training datasets for HLA-DR4(B1*0401) 462 Binding peptides and 177 non-binding peptides that have interacted with the HLA-DR4(B1*0401)constitute the HLA-DR4(B1*0401)training set.The binding peptides are extracted from SYFPEITHI[7],which have been described by Nielsen,etal[4];The nonbinders are extracted from MHCBN[8],which have been described by Murugan,etal[9].Both the training set and the evaluation set contain two columns.The first column gives the peptide sequence,and the second one gives theIC50logtransformed binding affinity pIC50,pIC50=1-log(IC50/(nmol·L-1))/log 50 000[4].This set is referred to as DR4-training.
1.1.2 Testing datasets for HLA-DR4(B1*0401) HLA-DR4(B1*0401)benchmarks are the same benchmarks used by Nielsen,etal[4].They consist of ten datasets;and eight of ten datasets are downloaded from MHCbench(http://www.imtech.res.in/raghava/mhcbench),the rest two are Southwood and Geluk datasets[4].The same threshold to determine binders and non-binders as Nielsen,etal.(2004)[4]is used in study in this paper.For the 8 MHCbench datasets,peptides with a binding value of non-zero are defined as binders and all other peptides are defined as non-binders.For the Southwood and Geluk datasets,an affinity of 1 000 nmol/L is taken as the threshold,which is peptides with an associated pIC50larger than 0.36 are defined as binding peptides.The 10 benchmarks are through homology reduction,which ensures that no peptide in the benchmarks has a match in the training set with more than 7 identical amino acids over an alignment of 9 amino acids.Tab.1 shows a summary of the original and the homology-reduced benchmark datasets,respectively.
1.1.3 IEDB HLA-DR restricted testing datasets A quantitative IEDB HLA-DR restricted peptide-binding data for each HLA-DR alleles partitioned into 5 datasets using the method described by Nielsen,etal[10].Each dataset and its corresponding partition are available online at http://www.cbs.dtu.dk/suppl/immunology/Net MHC-2.0.php.
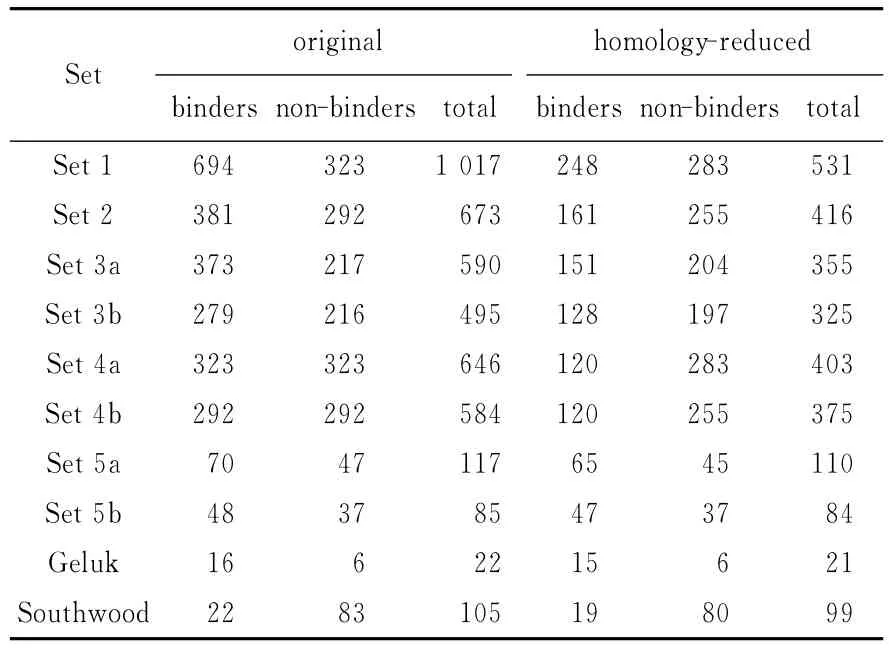
Tab.1 Description of HLA-DR4(B1*0401)testing datasets
1.2 AOG algorithm
1.2.1 Core nonamers filter There is a binding core in the binding peptides to MHCⅡmolecules,which is approximately 9 amino acids long.This binding core reveals some distinctions from randomness in the frequency of amino acids(i.e.,the background in the SWISS-PROT database[11]).And a statistically significant alignment is likely to grasp such distinctions[12].On the basis of this idea,the algorithm samples possibly ungapped alignment fromnpeptide sequences(nis the number of binding peptides in the training set).Because nearly all the binding peptides have a hydrophobic residue(F,I,L,M,V,W,Y)at P1 position[13],the sampling restricts to the ungapped nonamers that have a hydrophobic residue at P1 position.The size of the search space could be greatly reduced,e.g.,given a binding peptide′GNKLCALLYGDAEKP′,nonamers for selecting are ′LCALLYGDA′ and′LLYGDAEKP′,and the other candidates that do not have a hydrophobic residue at P1 are discarded.
1.2.2 Sequence weights Closely related sequences carry similar information,and a large set of them make the raw amino acid frequencies calculation badly biased.Hobohm 1-like algorithm[14]is used for clustering the sequences and a sequence identity of 62%is used as the cluster threshold,e.g.,if sequence A has 6(≥9×62%)amino acids identical to the sequence B in their aligned positions,A and B are clustered,and are assigned a weight 1/2.If C has 6 amino acids identical to sequence A or B in their aligned positions,A,B and C are clustered and assigned a weight 1/3.
1.2.3 Scoring matrix calculation Pseudofrequency method is used for estimating the frequency of amino acids for low counts[4].For an alignment,the pseudo-count frequency of amino acidiat positionjis
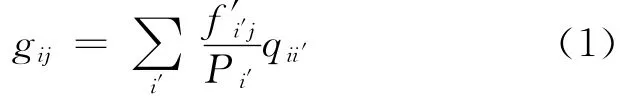
Wheref′i′jis the observed frequency of amino acidi′at positionj.Pi′is the background frequency of amino acidi′in the SWISS-PROT database[11].qii′is calculated as
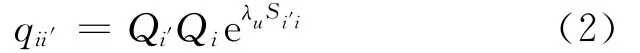
WhereQi′is the observed frequency of amino acidi′in the alignment andλuis a random scale number(2 by default);Si′iis the observed probability of occurrence foriandi′amino pair from the Blosum62 substitution matrix[14].
The effective amino acid frequency is
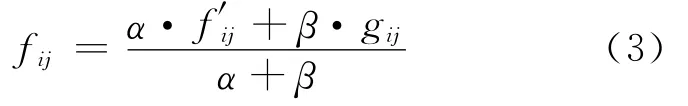
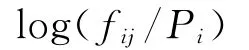
Whereαis the number of clusters,βis the weight on the pseudo-count correction.A too great value ofβwould reduce the sensitivity of scoring prediction matrix.The score of the amino acidiin positionjis computed as log-odds ratios: Since different positions have different impact on the binding peptides and MHCⅡmolecule interaction,i.e.,anchor positions are more important than ordinary positions.The positiondetermined parameterμjis introduced;And the final 9×20 scoring matrixMis calculated as
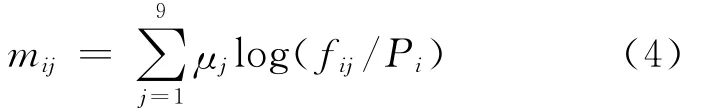
The score of a nonamer subpeptide is the sum of all the scores of amino acids in the nine positions.And the score of a peptide is the highest score of all nonamer subpeptides in the peptide sequence.
1.2.4 AUC calculation The receiver operator characteristics(ROC)curve is a twodimensional curve;the false positive rate of the prediction is plotted on theXaxis and the true positive rate of the prediction is plotted on theYaxis over a continuous range of cut-off values from high to low.The AUC value is the area under the ROC curve;it reflects the ability of a model that can tell a randomly chosen positive instance from a randomly chosen negative one[15].In the study,ROC analysis is used for measurement of the ability of different models to identify the MHC classⅡepitopes.Homology reduced AUC is calculated on the Hobohm 1-like[14]homology-reduced training dataset.This implement ensures that there are not two peptides in the training set that have over nine identical continuous amino acids.
1.2.5 AOG algorithm (i)raw alignment:a new starting point is chosen randomly in a peptide sequence.The random alignment is run for 5 000 times to reach a relatively high-AUC alignment.Since the alignment space has a very large number of local maxima with close to identical prediction accuracy,this procedure is repeated 100 times with different initial configurations.The probability of accepting a new alignment in the sampling is calculated as:
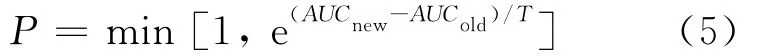
WhereTis a scalar.(ii)Precise alignment:for the starting point of the binding peptide voted by a majority of the 100 alignments in(i),twice selecting probabilities of other starting points are used.The precise alignment is run for 100 000 times to reach the final optimal alignment.(iii)The two factors that influence the performance of the scoring matrices are the weightβin the effective amino acid frequency calculation and the position specific weightμj.A two-stage Monte Carlo method[16]is implemented,alternately shiftingμjin Eq.(4)andβin Eq.(3)to optimize these parameters.
In(i),the scalarTimplicit in Eq.(5)is set to 0.001,that reduces the probability of accepting unfavorable alignment;In(ii),the scalarTis set from 0.1 to 0.001,that gradually reduces the probability of accepting unfavorable alignment;In(iii),the scalarTis set to 0.001,that reduces the probability of accepting unfavorable score matrices.The alteredTin(ii)makes the probabilityPunfixed and accordingly guarantees the alignment chain irreducible and aperiodic(and thus ergodic)[16].
2 Results
2.1 MHCⅡ(HLA-DR4(B1*0401))weight matrix extraction
Using HLA-DR4(B1*0401)training data DR4-training,an AUC-guided iterative training process is employed to get the optimal alignment,parameters and the corresponding scoring matrix.The final scoring matrix for HLA-DR4(B1*0401)is shown in Fig.1.Each itemmijof the scoring matrixMrespectively corresponds to a kind of amino acidiin a sequence positionj,and the sum of these scores is the predicted binding affinity.Hence,the scoring matrixMcan be seen as the impact of each amino acid in sequence positions on the binding affinity.The height of the symbol of the amino acidiis proportional to the absolute value ofmij.The upside or upside-down symbol represents the positive or negative sign ofmijrespectively.The colors of amino acid symbols represent their physicochemical characteristics,i.e.,black,neutral and hydrophobic;blue,basic;green,neutral and polar;red,acidic.(due to print limit,here using different shades for demonstration)

Fig.1 The weight coefficients of amino acids in HLA-DR4(B1*0401)peptides
Each symbol column corresponds to a sequence position between P1 and P9.
2.2 Results for the HLA-DR4(B1*0401)data
The performance of the AOG method,Gibbs sampling method and TEPITOPE are compared on the HLA-DR4(B1*0401)benchmarks.The results of Gibbs sampler are calculated with the weight matrix offered by Nielsen;this weight matrix is trained with the positive samples of the DR4-training;the results of TEPITOPE are gained with the weight matrix from ProPred[6].The AUC value of each method on the 10 benchmarks is illustrated in Fig.2(a)and Fig.2(b).It is observed that AOG gives a better performance than the Gibbs sampler and TEPITOPE.The average AUC values on the original and homology reduced benchmarks are 0.771 and 0.713,respectively.The average AUC values are 0.744 and 0.673 for the Gibbs sampler and 0.702 and 0.667 for TEPITOPE.
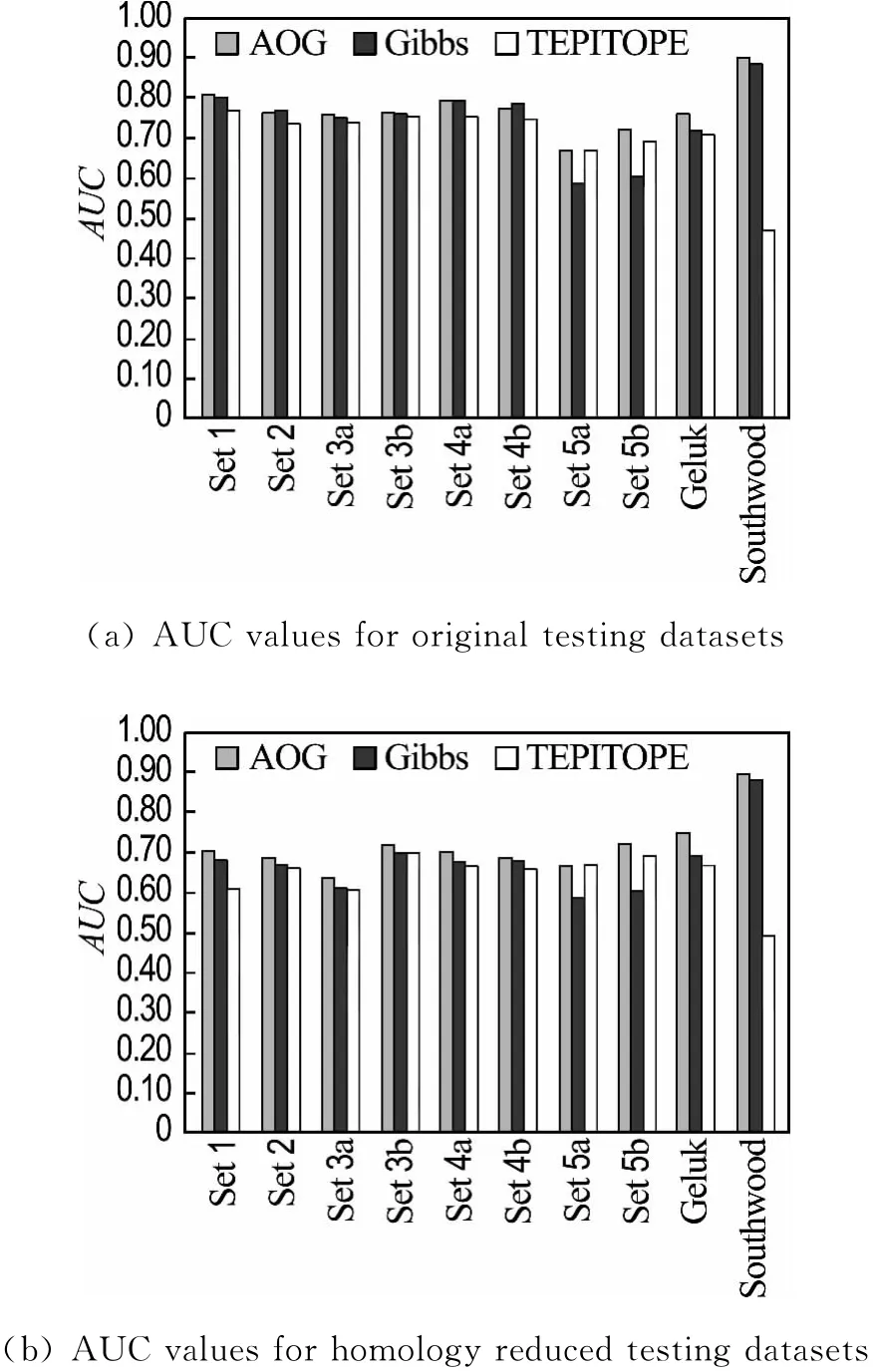
Fig.2 Prediction performance of various methods on the HLA-DR4(B1*0401)benchmarks
2.3 Results for quantitative IEDB HLA-DR data
The predictive performances of AOG and TEPITOPE on the quantitative IEDB benchmark datasets are estimated using five-fold crossvalidation.In each cross-validation,1/5 of the data are left out for evaluation and the remaining 4/5 are used for an alternating training.The predictive performances of AOG and TEPITOPE on the 11 HLA-DR allele benchmarks are shown in Tab.2.The predictive performances of AOG method and TEPITOPE are in terms of AUC values,using a binding affinity threshold of 500 nmol/L.The results of TEPITOPE are obtained with the use of the scoring matrix from ProPred[6].Since ProPred offers scoring matrices for only 11 alleles,the rest 3 alleles are not included in the table.It is clear that AOG method has a higher performance than TEPITOPE for most alleles(10/11).Only for one allele(DRB1*0404)does the TEPITOPE outperform AOG method.
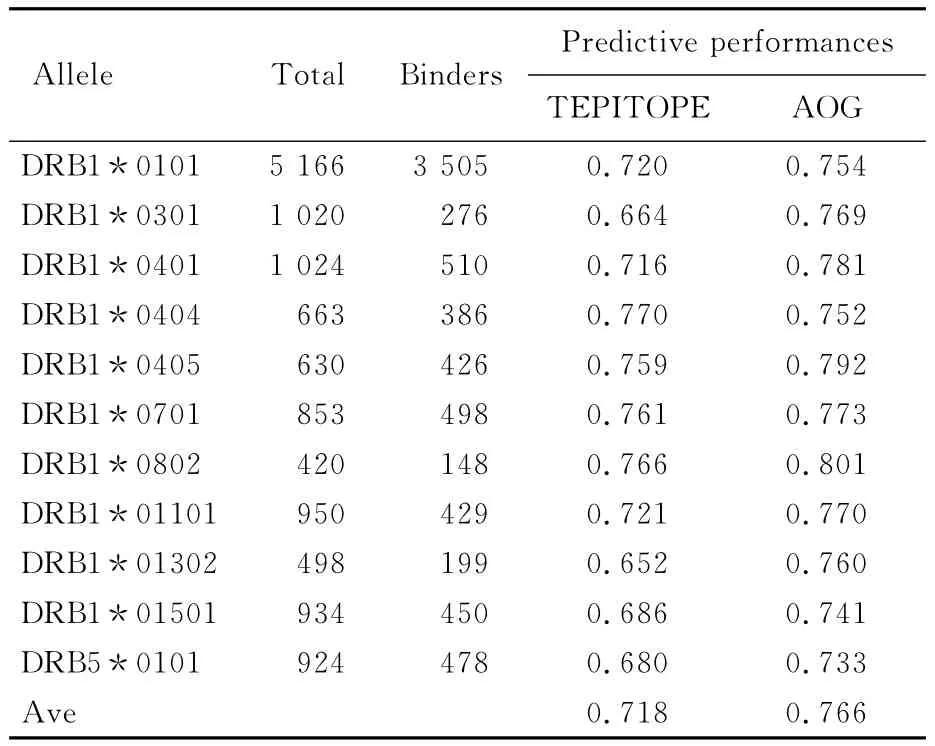
Tab.2 Predictive performances of AOG and TEPITOPE for the 11 HLA-DR alleles in the quantitative IEDB benchmark datasets
3 Discussion
As shown in Fig.1,the height of each amino acid symbol is proportional to its absolute score,which is the contribution of the amino acid in a sequence position to the MHC-peptide binding affinity,and the height of all amino acid symbols stacked on each position along P1-P9 is proportional to the sum of corresponding absolute scores for the 20 possible amino acids on the position,which is the contribution to the binding affinity.The positions in core region of the peptides have distinct specificities,i.e.,P1,P4,P6 and P9 positions have distinct influence on the HLAⅡ-peptide binding.
For the purpose of interpreting the amino acid characteristics in HLAⅡ-peptide primary positions that are presented in Fig.1,the crystal structures of DRB1*0401(PDB id:1J8 H,1D5Z,1D6E,2SEB,1D5M,1D5X)are used to find amino acids that make nonbonding contact with the peptides(i.e.,two residues are defined to be nonbonding contact if the distance of two atoms of these residues is smaller than 0.4 nm)[17].Tab.3 lists the amino acids of α-chain andβ-chain of DRB1*0401 nonbonding contact with the peptides.

Tab.3 The amino acids of the HLA-DR4(B1*0401)molecule nonbonding contact with the peptides
Neutral amino acids are colored black,electropositive and basic amino acids are colored blue,and electronegative and acidic amino acids are colored red(due to print limit,here using different shades for demonstration).
P1 position:Residues of the HLA DRB1*0401 molecule that make nonbonding contact with residues of peptides in P1 position are Ile7,Phe24,Ile31,Phe32,Trp43,Ala52,Ser53,Phe54 in theα-chain and Asn82,Val85,Gly86,Phe89 in theβ-chain(see Tab.3).It is found that,the P1 pocket is shaped by conserved aliphatic amino acids(Ileα7,Ileα31,Serα53, Alaα52,Valβ85,Glyβ86)and aromatic amino acids(Pheα24,Pheα32,Pheα54,Trpα43,Pheβ89)and represents a highly hydrophobic environment.Jardetzky′s single residue substitution experiment[13]demonstrates that the main determinant of binding is a large pocket that accommodates a hydrophobic or aromatic amino acid side chain near the N terminus of the peptide(the P1 position).From Fig.1,the following can be figured out:(i)The sum of absolute values in P1 position is significantly larger than that of any other position,indicating that P1 position has the highest influence on the binding affinity;(ii)Polar amino acids have negative scores in P1,which indicates that polar amino acids are unfavorable to the binding;Hydrophobic amino acids have positive scores in P1,which indicates that these hydrophobic amino acids are favorable to the binding;(i)and(ii)are in accordance with the Jardetzky′s conclusion[13].(iii)Stable amino acid residues F,W and Y have much higher scores than less stable amino acid residues I,L,M and V,which indicates that stable amino acid residues are more favorable to the binding;this result is in accordance with the Tobita′s conclusion[18]—Difference in stability of amino acids in P1 closely correlates with the binding affinity.
P4 position:Residues of the HLA DRB1*0401 molecule that make nonbonding contact with residues of peptides in P4 position are Gln9,Asn62 in theα-chain and His13,Phe26,Asp28,Gln70,Lys71,Ala74,Tyr78 in theβchain(Tab.3).The electronegative and acidic amino acids Aspβ28 and electropositive and basic amino acids Hisβ13 and Lysβ71 endow the P4 pocket polar binding characteristics;Previous studies[19]on the HLA-peptide affinity have shown that the positively charged Lysβ71 can make direct contact with side-chain residues from the antigenic peptide;Lysβ71 makes this pocket tend to have a high affinity for negatively charged or uncharged polar amino acids,whereas disfavors positively charged amino acids(like Lys).This is an approval of the algorithm in P4 position:in Fig.1,negatively charged amino acids Asp and Glu have the highest positive scores,whereas electropositive and basic amino acids Lys,Arg and His have the lowest negative scores.From Fig.1,it is also found that bulky amino acids Phe and Trp also have relatively high positive score that may indicate that P4 pocket is a large sized one.
P6 position:Residues of the HLA DRB1*0401 molecule that make nonbonding contact with residues of peptides in P6 position are Glu11,Asn62,Val65,Asp66,Asn69 in theαchain and Val11,His13 and Lys71 in theβ-chain(Tab.3).As indicated by the amino acid symbol height stacked in P6 position in Fig.1,P6 is a major anchor and inhibitory residue position.The electronegative and acidic amino acids Gluα11,Aspα66 and electropositive and basic amino acids Hisβ13 and Lysβ71 endow the P6 pocket a polar interface;The experimental results[20]have shown that this pocket favors the binding of medium sized(like Met,Leu and Ile)or polar amino acid residues.In the scoring matrix(Fig.1),the negatively charged amino acids Asp and polar amino acids Asn,Ser and Thr have positive scores,which may be beneficial to the binding affinity.
P9 position:Residues of the HLA DRB1*0401 molecule that make nonbonding contact with residues of peptides in P9 position are Asn69,Ile72,Met73,Arg76 in theα-chain and Glu9,Tyr37,Asp57,Tyr60,Trp61 in theβchain(Tab.3).As indicated by the amino acid symbol height stacked in P9 position in Fig.1,P9 is a major anchor and inhibitory residue position.The P9 pocket is shaped by neutral amino acids Tyrβ37 and Tyrβ60 and electronegative and acidic amino acids Gluβ9 and Aspβ57[21].So the positively charged or polar residues are favored in the P9 pocket.In the scoring matrix(Fig.1),the positively charged amino acids His and polar amino acids Gly,Ser and Gln have positive scores.It is indicated that these amino acids may enter the inner cavity wall of P9 easily.As an unexpected result,the hydrophobic amino acid Ala has the highest score in the P9 position,which indicates that P9 pocket is a small sized pocket.
4 Conclusion
A method,AUC Optimized Gibbs(AOG)is developed for prediction of peptide binding to MHCⅡmolecules.Tests on 10 HLA-DR4(B1*0401)benchmarks and quantitative IEDB HLA-DR benchmark show that AOG is a better predictive method for MHC classⅡepitopes than Gibbs sampling method and TEPITOPE.The positions in core region of the HLA-DR4(B1*0401)peptides have distinct specificities,i.e.,P1,P4,P6 and P9 positions have distinct influence on the MHC-peptide binding.
[1]Parker K C,Bednarek M A,Coligan J E.Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains[J].Journal of Immunology,1994,152(1):163-175.
[2]Doytchinova I A,Blythe M J,Flower D R.Additive method for the prediction of proteinpeptide binding affinity.Application to the MHC class I molecule HLA-A*0201[J].Journal of Proteome Research,2002,1(3):263-272.
[3]Peters B,Sette A.Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method[J].BMC Bioinformatics,2005,6:132.
[4]Nielsen M,Lundegaard C,Worning P,etal.Improved prediction of MHC class I and classⅡepitopes using a novel Gibbs sampling approach[J].Bioinformatics,2004,20(9):1388-1397.
[5]Nielsen M,Lundegaard C,Lund O.Prediction of MHC classⅡbinding affinity using SMM-align,a novel stabilization matrix alignment method[J].BMC Bioinformatics,2007,8:238.
[6]Sturniolo T,Bono E,Ding J,etal.Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA classⅡmatrices[J].Nature Biotechnology,1999,17(6):555-561.
[7]Rammensee H,Bachmann J,Emmerich N P,etal.SYFPEITHI:database for MHC ligands and peptide motifs[J].Immunogenetics,1999,50(3-4):213-219.
[8]Bhasin M,Singh H,Raghava G P.MHCBN:a comprehensive database of MHC binding and nonbinding peptides[J].Bioinformatics,2003,19(5):665-666.
[9]Murugan N,Dai Y.Prediction of MHC classⅡbinding peptides based on an iterative learning model[J].Immunome Research,2005,1:6.
[10]Nielsen M,Lundegaard C,Blicher T,etal.Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence:Net MHCⅡpan[J].PLoS Computational Biology,2008,4(7):e1000107.
[11]Bairoch A,Apweiler R.The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000[J].Nucleic Acids Research,2000,28(1):45-48.
[12]Altschul S F,Madden T L,Schffer A A,etal.Gapped BLAST and PSI-BLAST:a new generation of protein database search programs[J].Nucleic Acids Research,1997,25(17):3389-3402.
[13]Jardetzky T S,Gorga J C,Busch R,etal.Peptide binding to HLA-DR1:a peptide with most residues substituted to alanine retains MHC binding[J].The EMBO Journal,1990,9(6):1797-1803.
[14]Hobohm U,Scharf M,Schneider R,etal.Selection of representative protein datasets[J].Protein Science:a Publication of the Protein Society, 1992,1(3):409-417.
[15]Swets J A.Measuring the accuracy of diagnostic systems[J].Science,1988,240(4857):1285-1293.
[16]Robert C P,Casella G.Monte Carlo Statistical Methods[M].2nd ed.New York:Springer,2004:267-280.
[17]Holzhütter H G,Kloetzel P M.A kinetic model of vertebrate 20S proteasome accounting for the generation of major proteolytic fragments from oligomeric peptide substrates[J].Biophysical Journal,2000,79(3):1196-1205.
[18]Tobita T,Oda M,Morii H,etal.A role for the P1 anchor residue in the thermal stability of MHC classⅡmolecule I-Ab[J].Immunology Letters,2003,85(1):47-52.
[19]Hill J A,Southwood S,Sette A,etal.Cutting edge:the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC classⅡmolecule[J].Journal of Immunology,2003,171(2):538-541.
[20]Zarour H M,Storkus W J,Brusic V,etal.NYESO-1 encodes DRB1*0401-restricted epitopes recognized by melanoma-reactive CD4+T cells[J].Cancer Research,2000,60(17):4946-4952.
[21]Atanasova M,Dimitrov I,Flower D R,etal.MHC classⅡbinding prediction by molecular docking[J].Molecular Informatics,2011,30(4):368-375.
