UPLC法同时测定盐酸阿糖胞苷原料药的含量和有关物质
2014-02-03海丽萍杨东菁王志远王雪芹河南省直属机关第一门诊部郑州45000河南大学医学院河南开封475001河南大学药学院河南开封475001河南省食品药品检验所郑州45000
海丽萍,杨东菁,王志远,王雪芹(1.河南省直属机关第一门诊部,郑州 45000;.河南大学医学院,河南开封 475001;.河南大学药学院,河南开封 475001;4.河南省食品药品检验所,郑州 45000)
UPLC法同时测定盐酸阿糖胞苷原料药的含量和有关物质
海丽萍1*,杨东菁2,王志远3,王雪芹4#(1.河南省直属机关第一门诊部,郑州 450003;2.河南大学医学院,河南开封 475001;3.河南大学药学院,河南开封 475001;4.河南省食品药品检验所,郑州 450003)
目的:建立同时测定盐酸阿糖胞苷原料药的含量和有关物质的方法。方法:采用超高效液相色谱法。色谱柱为Inertsil ODS-3C18,流动相为磷酸盐缓冲液-甲醇(梯度洗脱),流速为0.8m l/min,检测波长为254 nm,柱温为40℃,进样量为10μl。结果:尿嘧啶、尿苷、阿糖尿苷、盐酸阿糖胞苷检测质量浓度分别在0.100 8~20.16、0.1~20.12、0.095 6~19.12、0.1~20.004μg/m l范围内与各自峰面积积分值呈良好的线性关系(r=0.999 9、0.999 8、0.999 9、0.999 9);精密度、稳定性、重复性试验的RSD≤0.79%;尿嘧啶、尿苷、阿糖尿苷平均加样回收率为103.8%、102.2%、99.7%,RSD分别为2.44%、2.69%、3.16%(n=9)。结论:该方法准确、灵敏度高、专属性强、重复性好,可用于盐酸阿糖胞苷原料药的质量控制。
盐酸阿糖胞苷;尿嘧啶;尿苷;阿糖尿苷;超高效液相色谱法;含量测定
*主管药师。研究方向:医院药学。电话:0371-65907512
#通信作者:主任药师,硕士。研究方向:药物分析。电话:0371-63388295。E-mail:wangxueqin2008@126.com
阿糖胞苷为嘧啶类抗代谢药,主要用于治疗急性粒细胞白血病及消化道肿瘤,是治疗白血病最常见的药物[1]。盐酸阿糖胞苷为阿糖胞苷的盐酸盐。美国药典(USP)、英国药典(BP)等均只收载阿糖胞苷原料药的检测质量标准[2-3],而《中国药典》2010年版收载了盐酸阿糖胞苷(原料药)的检测质量标准[4]。BP收载的有关物质测定采用薄层色谱(TLC)法,含量测定采用非水滴定法;USP与《中国药典》收载的有关物质和含量测定均采用高效液相色谱(HPLC)法,但色谱条件有所不同。笔者参照了相关文献[5-9],采用超高效液相色谱(UPLC)法同时对盐酸阿糖胞苷原料药中主成分和有关物质的含量进行测定,以为其质量控制提供参考。
1 材料
Acquity系列UPLC仪,包括四元梯度泵、自动进样器、光电二极管阵列检测器、Empower工作站(美国Waters公司);XR205SM-DR电子天平(瑞士Precisa公司)。
盐酸阿糖胞苷、阿糖尿苷对照品(美国Sigma公司,批号:SLBB7397V、031M 5069V);尿嘧啶、尿苷对照品(中国食品药品检定研究院,批号:100469-200401、887-200202);盐酸阿糖胞苷原料药(河南信心制药有限公司,批号:20120401、20120402、20120403、20120801、20120802、20120803);甲醇为色谱纯,磷酸二氢钠、磷酸氢二钠、磷酸均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:InertsilODS-3C18(4.6mm×150mm,5μm);流动相:磷酸盐缓冲液(流动相A,含0.01mol/L磷酸二氢钠和0.01 mol/L磷酸氢二钠)-甲醇(流动相B),梯度洗脱程序见表1;流速:0.8m l/min;检测波长:254 nm;柱温:40℃,进样量:10μl。
2.2 溶液的制备


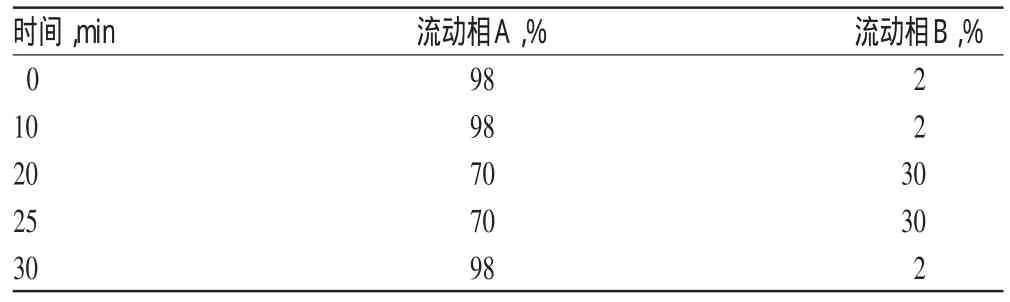
表1 流动相梯度洗脱程序Tab 1 Gradient elution ofmobile phase
2.2.3供试品溶液的制备 取样品0.05 g,置于10m l量瓶中,加水稀释至刻度,摇匀,即得供试品溶液。
2.2.4 空白对照溶液的制备 取不含盐酸阿糖胞苷的样品,按“2.2.3”项下方法制成空白对照溶液。
2.3 专属性试验
2.3.1 主成分含量测定的专属性考察 分别取“2.2”项下的对照品溶液、供试品溶液和空白对照溶液各10μl,分别注入UPLC仪,记录色谱,详见图1。由图1可见,空白对照对盐酸阿糖胞苷含量测定无干扰。
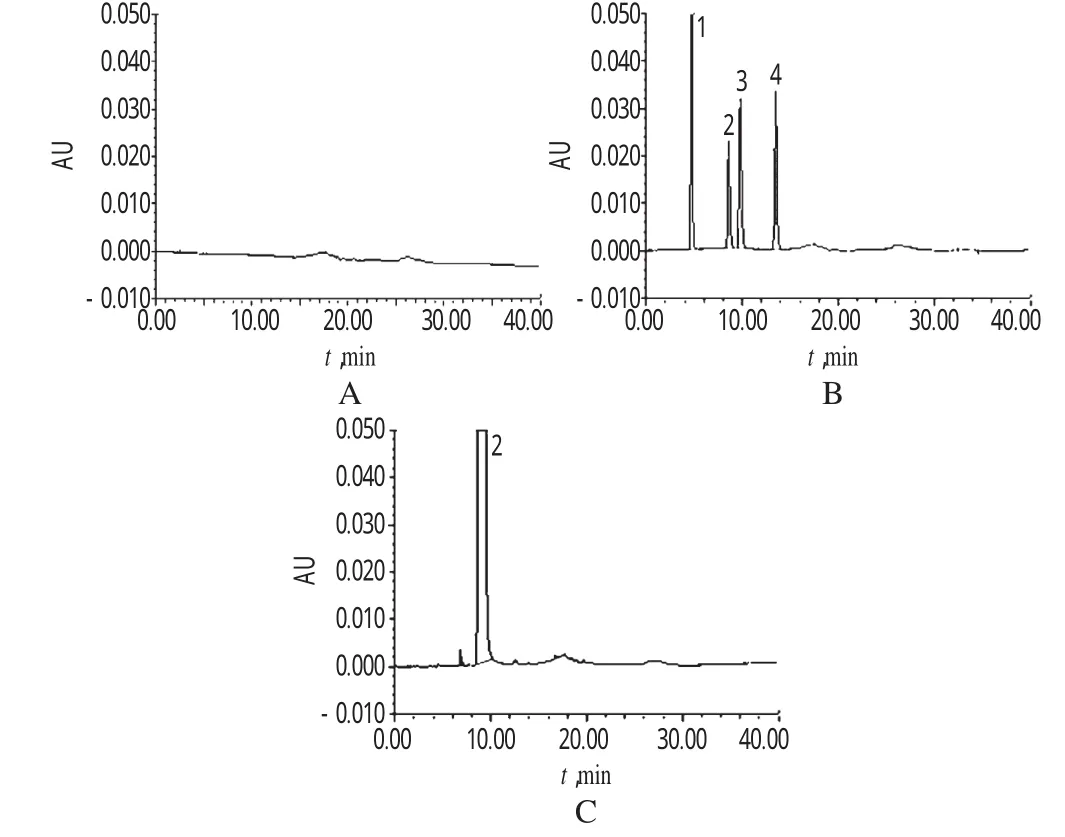
图1 超高效液相色谱图A.空白对照;B.对照品;C.供试品;1.尿嘧啶;2.盐酸阿糖胞苷;3.尿苷;4.阿糖尿苷Fig 1 UPLC chromatogram sA.blank control;B.substance control;C.test sample;1.uracil;2. cytarabine hydrochloride;3.uridine hydrochloride;4.1-β-D-arabino furanosyl-uracil
2.3.2破坏性试验(1)精密称取样品0.005 2 g,置于10m l量瓶中,加1mol/L盐酸溶液1m l,室温放置0.5 h,用1mol/L氢氧化钠溶液中和至中性,加水稀释至刻度,摇匀,备用;(2)精密称取样品0.005 5 g,置于10m l量瓶中,加1mol/L氢氧化钠1m l,室温放置0.5 h,用1mol/L盐酸溶液中和至中性,加水稀释至刻度,摇匀,备用;(3)精密称取样品0.005 3 g,置于10m l量瓶中,加30%过氧化氢1m l,室温放置0.5 h,加水稀释至刻度,摇匀,备用;(4)精密称取样品0.005 2 g,于105℃干燥6 h,置于10m l量瓶中,加水稀释至刻度,摇匀,备用;(5)精密称取样品0.005 1 g,于(4 500±500)lx条件下照射8 h,置于10m l量瓶中,加水稀释至刻度,摇匀,备用。精密量取上述5种溶液各10μl,分别注入UPLC仪,记录色谱,详见图2。由图2可见,本品在强酸、强碱、氧化、高温、光照条件下,样品均被破坏产生降解产物,所产生的降解产物在“2.1”项色谱条件下均能达到较好的分离。主峰与降解产物峰、各个杂质峰之间分离度均大于1.5。
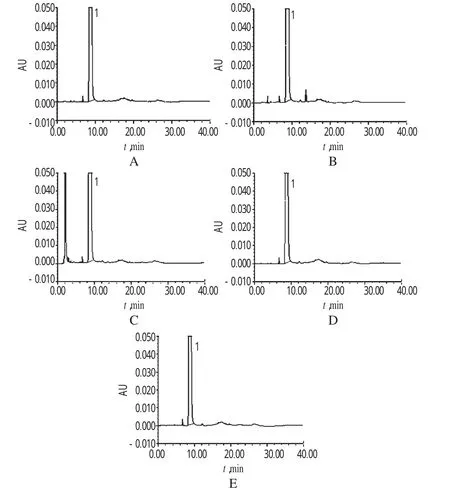
图2 破坏性试验超高效液相色谱图A.酸破坏样品;B.碱破坏样品;C.氧化破坏样品;D.高温破坏样品;E.光照破坏样品;1.盐酸阿糖胞苷Fig 2 HPLC chromatogram s of treated testA.treated w ith acid;B.treated w ith alkali;C.treated w ith oxidation;D.treated w ith high temperature;E.treated w ith light;1.cytarabine hydrochloride
2.4 线性关系考察
取“2.2.1”项下4种对照品贮备液各0.4、0.2、0.1、0.05m l,分别置于10m l量瓶中,加水稀释至刻度,摇匀。按“2.1”项下色谱条件,分别取上述4种溶液10、5、3、2、1、0.5、0.3、0.1μl进样,记录色谱。以检测质量浓度(x,μg/m l)为横坐标,峰面积(y)为纵坐标,进行线性回归,回归方程和线性范围见表2。

表2 回归方程和线性范围Tab 2 Regression equation and linear range
2.5 检测限和定量限
取“2.2.2”项下对照品溶液适量,按“2.1”项下色谱条件进样,记录色谱,以信噪比S/N=3∶1计算盐酸阿糖胞苷、尿嘧啶、尿苷、阿糖尿苷的检测限,分别为3.00、1.008、3.018、2.868 ng;以信噪比S/N=10∶1计算盐酸阿糖胞苷、尿嘧啶、尿苷、阿糖尿苷的定量限,分别为10.00、3.024、5.03、4.78 ng。
2.6 精密度试验
取“2.2.2”项下对照品溶液10μl,按“2.1”项下色谱条件连续进样测定6次,结果,尿嘧啶、尿苷、阿糖尿苷、盐酸阿糖胞苷的RSD分别为0.08%、0.28%、0.28%、0.54%,表明仪器精密度良好。
2.7 稳定性试验
取“2.2.2”项下对照品溶液适量,分别于放置0、1、2、3、5、6、10、15 h时进样测定。结果,尿嘧啶、尿苷、阿糖尿苷、盐酸阿糖胞苷的RSD分别为0.07%、0.25%、0.37%、0.65%,表明15 h内对照品溶液稳定性良好。
2.8 重复性试验
取样品(批号:20120401)适量,共6份,按“2.2.3”项下方法制备供试品溶液,并按“2.1”项下色谱条件进样测定并计算各成分含量。结果,尿嘧啶、尿苷、阿糖尿苷均未检出,其它单杂(胞苷)的量平均为0.002%,RSD=0.79%,表明本方法重复性良好。
2.9 有关物质加样回收率试验
精密称取样品(批号:20120401)共9份,每份约0.05 g,分别置于10m l量瓶中,精密加入“2.2.1”项下对照品溶液适量,加水稀释至刻度,摇匀,按“2.2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定并计算加样回收率,结果见表3。

表3 有关物质加样回收率试验结果(n=9)Tab 3 The average recoveries of related substances(n=9)
2.10 样品含量测定
取6批样品适量,按“2.2.3”项下方法制备供试品溶液,并按“2.1”项下色谱条件进样测定,记录色谱,按外标法以峰面积计算含量,结果见表4。

表4 样品含量测定结果(%,n=3)Tab 4 Results of content determ ination of sam p les(%,n=3)
2.11 相对保留时间及相对校正因子的测定

2.12 样品有关物质测定
精密称取样品50mg,置于10m l量瓶中,加水溶解并稀释成每1m l含5mg的溶液,作为有关物质测定供试品溶液。取盐酸阿糖胞苷对照品适量,精密称定,用水溶解并定量稀释制成每1m l含5μg的溶液作为有关物质测定对照品溶液。按“2.1”项下色谱条件,取上述对照品溶液10μl,注入UPLC仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。再精密量取上述供试品溶液与对照品溶液各10μl,分别注入UPLC仪,测定并计算盐酸阿糖胞苷的含量。结果,尿嘧啶峰、尿苷峰、阿糖尿苷峰对盐酸阿糖胞苷峰的相对保留时间分别为0.53、1.14、1.55,尿嘧啶峰、尿苷峰、阿糖尿苷峰对盐酸阿糖胞苷峰的相对校正因子分别为2.92、1.73、1.55;对盐酸阿糖胞苷峰的相对保留时间约为0.38、0.43的色谱峰的相对校正因子均为1.72,其他杂质峰的相对校正因子均按1.1计算。结果见表5。
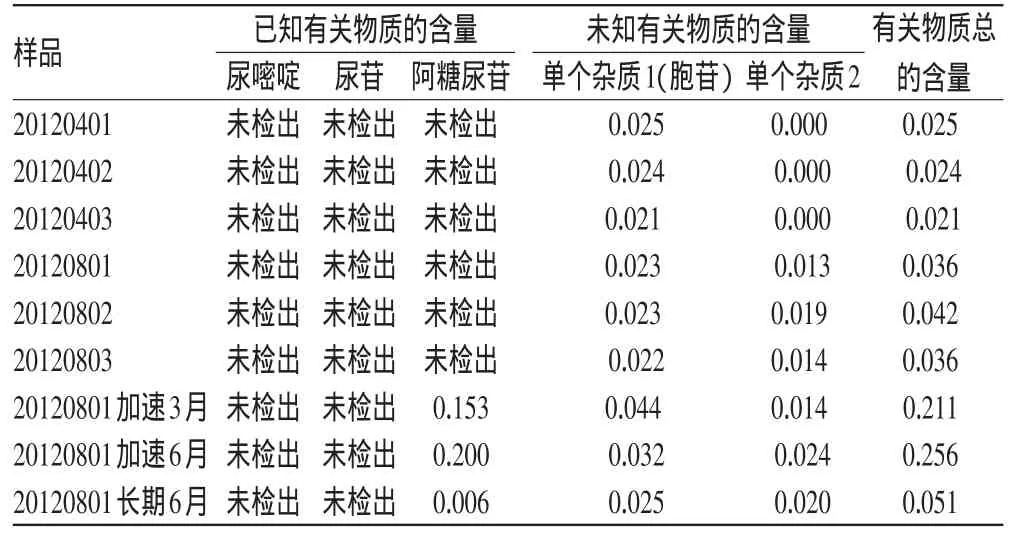
表5 样品有关物质测定结果(%)Tab 5 Results of content determ ination of related substances in sam p les(%)
3 讨论
3.1 检测波长的选择
色谱图中可见尿嘧啶、尿苷、阿糖尿苷及盐酸阿糖胞苷的最大吸收波长分别为259.9、262.1、263.3和272.5 nm,但由于盐酸阿糖胞苷在254 nm波长处的吸收较强,既可以保证杂质的检出,又能够准确地测定主成分含量,故选择254 nm作为本方法的检测波长。
3.2 流动相的选择
笔者研究发现,流动相中0.01mol/L磷酸氢二钠和0.01 mol/L磷酸二氢钠溶液的pH均为6.86,缓冲力较大,且较稳定。若将pH调至6.0或7.0,则需加入大量的稀酸或稀碱,不易控制,色谱峰易分叉。另外,由于本品黏度较大,重复多次进样后色谱柱上残留物不易洗脱干净,柱压不断升高,因此在梯度洗脱主成分和主要杂质出峰后,增加了甲醇的比例,有利于洗脱色谱系统中残留杂质,保持色谱条件的稳定。
3.3 色谱柱的选择
笔者曾尝试采用了3根不同品牌的C18色谱柱、两台不同型号的UPLC仪,分别在不同时间点采用本研究中的色谱条件进行测定。结果发现,Inertsil ODS-3 C18分离效果最好,无干扰,灵敏度高,且峰形较好。
3.4 样品的稳定性
在破坏性试验中均检出了尿嘧啶和尿苷,加速试验和长期试验中均检出了阿糖尿苷。盐酸阿糖胞苷对温度较敏感,可在放置过程中降解产生阿糖尿苷。因此,在贮藏时应注意温度对样品质量的影响。
综上所述,本方法准确、灵敏度高、专属性强、重复性好,可作为盐酸阿糖胞苷原料药的质量控制方法。
[1] 李莉霞,唐跃年.阿糖胞苷抗白血病药理作用及耐药机制的研究进展[J].中国药房,2006,17(19):1 506.
[2]British Pharmacopoeia Comm ission.BP2013[S].2012:631.
[3]The United States Pharmacopoeia Convention.USP35-NF30[S].2012:2 800.
[4]国家药典委员会.中华人民共和国药典:二部[S.]2010年版.北京:中国医药科技出版社,2010:716.
[5]左英熹,李良,卢炜,等.高效液相色谱法测定体内脱氧核糖核酸结合的阿糖胞苷浓度[J].中国医院药学杂志,2008,28(9):718.
[6]张虹,方昱,李英,等.高效液相色谱法测定细胞内三磷酸阿糖胞苷的浓度[J].中国医院药学杂志,2010,30(18):1 564.
[7] 梁蔚阳.高效液相色谱法测定阿糖胞苷注射液中有关物质的含量[J].药物生物技术,2009,16(5):460.
[8]林焕泽,蓝忠,杨华,等.反相高效液相色谱法测定注射用盐酸阿糖胞苷的有关物质[J].中国药业,2009,18(15):27.
[9]许双临,陈子春,林宇涵,等.大剂量阿糖胞苷治疗时血浆阿糖胞苷及阿糖尿苷的HPLC测定方法.[J].海峡药学,2013,25(7):128.
Simultaneous Determ ination of Cytarabine Hydrochloride and Related Substances by UPLC
HAILi-ping1,YANG Dong-jing2,WANG Zhi-yuan3,WANG Xue-qin4(1.First Outpatient Department,Departments Directly under Henan Province,Zhengzhou 450003,China;2.Medical College of Henan University,Henan Kaifeng 475001,China;3.Pharmaceutical College of Henan University,Henan Kaifeng 475001,China;4.Henan Institute for Food and Drug Control,Zhengzhou 450003,China)
OBJECTIVE:To establish a method for simultaneous determination of cytarabine hydrochloride and related substance.METHODS:UPLCmethod was adopted.The determination was performed on Inertsil ODS-3 C18column w ithmobile phase consisted of phosphate buffer-methanol(gradient elution)at the flow rate of 0.8 m l/min;the detection wavelength was set at 254 nm and the column temperature was 40℃.The sample size was 10μl.RESULTS:The liner ranges were 0.100 8-20.16μg/m l for uracil(r=0.999 9),0.1-20.12μg/m l for uridine(r=0.999 8),0.095 6-19.12μg/m l for 1-β-D-arabino furanosyl-uracil(r=0.999 9)and 0.1-20.004μg/m l for cytarabine hydrochloride(r=0.999 9),respectively.RSD of precision,stability and reproducibility tests were all lower than 0.79%;average recoveries of uracil,w ridine and 1-β-D-arabino furanosyl-uracil were 103.8%(RSD=2.44%,n=9),102.2%(RSD=2.69%,n=9)and 99.7%(RSD=3.16%,n=9).CONCLUSIONS:Themethod is accurate,sensitive,specific and reproducible,and can be used for the quality control of cytarabine hydrochloride.
Cytarabine hydrochloride;Uracil;Uridine;1-β-D-arabino furanosyl-uracil;UPLC;Content determination
R917
A
1001-0408(2014)12-1137-04
DOI 10.6039/j.issn.1001-0408.2014.12.28
2013-12-14
2014-1-13)
·药学教育·
