毛细管电泳法同时测定清肺抑火丸中5种蒽醌类成分的含量Δ
2014-02-03王晓可毋福海广东药学院广州510224
陈 岑,王晓可,毋福海(广东药学院,广州 510224)
毛细管电泳法同时测定清肺抑火丸中5种蒽醌类成分的含量Δ
陈 岑*,王晓可,毋福海#(广东药学院,广州 510224)
目的:建立同时测定清肺抑火丸中5种蒽醌类成分大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚含量的方法。方法:采用毛细管电泳法,以对乙酰氨基酚为内标。毛细管柱为未涂层弹性融硅石英毛细管柱,运行缓冲液为25mmol/L硼砂溶液+20 mmol/L十二烷基磺酸钠+4mmol/L磺丁基醚-β-环糊精+2%甲醇(pH 10.01),分离电压为18 kV,进样方式为重力进样10 s(高度15 cm),检测波长为254 nm,温度为25℃,湿度为<70%。结果:大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚检测质量浓度分别在5.0~40.0、2.4~19.2、3.6~28.8、11.2~89.9、2.5~20.0μg/m l范围内同各自峰面积与内标峰面积比值呈良好的线性关系(r=0.998 0、0.997 1、0.996 9、0.998 5、0.998 3);精密度、稳定性、重复性试验的RSD≤2.40%;平均加样回收率分别为96.6%、99.6%、99.2%、98.2%、98.6%,RSD分别为0.80%、1.32%、2.21%、1.63%、2.02%(n=6)。结论:该方法专属性强,结果准确、可靠,重复性好,可用于清肺抑火丸的质量控制。
毛细管电泳法;清肺抑火丸;大黄素;芦荟大黄素;大黄酸;大黄酚;大黄素甲醚;含量测定
*硕士研究生。研究方向:现代分析方法在中药质量控制中的应用。E-mail:chencen30603086@163.com
#通信作者:教授,博士。研究方向:现代分析方法在中药质量控制中的应用。E-mail:fuhaiwu@163.com
清肺抑火丸是由大黄、黄芩、黄柏、栀子、知母、浙贝母、桔梗、苦参、前胡和天花粉10味中药制成的中药成方制剂,具有清肺止咳、化痰通便之功效,临床用于治疗痰热阻肺所致的咳嗽、痰黄稠黏、口干咽痛、大便干燥,以及急性上呼吸道感染、支气管炎、咽炎、肺炎等。该药现行标准采用高效液相色谱(HPLC)法测定黄芩苷含量[1]。曾有文献报道采用薄层扫描法测定该药中盐酸小檗碱和菝葜皂苷元的含量[2];采用HPLC法测定该药中白花前胡甲素、盐酸小檗碱、芒果苷和大黄酸、大黄素等4种蒽醌类成分含量[3-5],以及黄芩苷等3种苷类成分含量[6]。而采用毛细管电泳法进行含量测定的研究较少见文献报道。大黄为清肺抑火丸方中主药,其化学成分主要有蒽醌类、大黄蒽类衍生物与葡萄糖结合成的苷类和蒽酮类如番泻苷A、B、C、D等,以及其他苷类、鞣质、多糖化合物、有机化合物和无机化合物等。其中,蒽类衍生物是大黄中主要的活性成分,而蒽醌类成分大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚一般作为大黄药材及其制剂质量控制的指标性成分。本研究建立了同时测定清肺抑火丸中大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚含量的毛细管电泳法,为清肺抑火丸的质量控制提供了新的依据。
1 材料
1.1 仪器
CL1030型高效毛细管电泳仪(北京彩陆科学仪器有限公司),配备K-2501型紫外-可见检测器(德国Knauer公司)、HW-2000型色谱工作站2.17版(南京千谱软件有限公司);ORION MODEL 828型pH计(美国Orion公司);KQ-400KDB型高功率数控超声波清洗器(昆山市超声仪器有限公司);HA-202M型电子天平(日本AND公司)。
1.2 药品与试剂
大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚、对乙酰氨基酚对照品(中国食品药品检定研究院,供含量测定用,批号:110756-200110、110795-200806、110757-200206、110796-201017、110758-200912、100018-200408);清肺抑火丸(甘肃佛仁制药科技有限公司,批号:20090502、20090503、20100702,规格:6 g/袋);磺丁基醚-β-环糊精(磺丁基醚-β-CD,山东新大精细化工有限公司,批号:080101,质量分数:99%);十二烷基磺酸钠(SDS)等试剂均为分析纯,水为双蒸水。
2 方法与结果
2.1 电泳条件
毛细管柱:未涂层弹性融硅石英毛细管柱(67 cm×75µm ID,有效长度60 cm);运行缓冲液:25mmol/L硼砂溶液+20 mmol/L SDS+4mmol/L磺丁基醚-β-CD+2%甲醇(pH 10.01);分离电压:18 kV;进样方式:重力进样10 s(高度15 cm);检测波长:254 nm;温度:25℃;湿度:<70%。毛细管柱在使用前依次用0.1mol/L氢氧化钠溶液、水、运行缓冲液各冲洗约5min。
2.2 溶液的制备
2.2.1 内标贮备液 精密称取对乙酰氨基酚对照品适量,加甲醇溶解稀释制成250μg/m l的内标贮备液。
2.2.2 对照品溶液 精密称取大黄素、大黄酚对照品12.5、21.8mg,各置于50m l量瓶中,加甲醇溶解并定容,摇匀,得质量浓度分别为250、562μg/m l的大黄素、大黄酚对照品溶液;精密称取芦荟大黄素、大黄酸、大黄素甲醚对照品12.1、18.1、12.6mg,分别置于100m l量瓶中,加甲醇溶解并定容,摇匀,得质量浓度分别为121、181、126μg/m l的芦荟大黄素、大黄酸、大黄素甲醚对照品溶液。
2.2.3 供试品溶液 取样品适量,研碎,取约4.0 g,精密称定,置100m l锥形瓶中,加2.5mol/L硫酸10m l,超声处理(功率:250W,频率:40 kHz)5m in,再加入三氯甲烷30m l,于70℃水浴回流1 h,冷却,移至分液漏斗中,分取三氯甲烷层,挥干三氯甲烷,残渣加乙醇20m l溶解,超声处理(功率:250W,频率:40 kHz)30min,滤过,水浴挥干乙醇,残渣加甲醇溶解至25m l量瓶中,加入2.5m l内标贮备液,加甲醇稀释至刻度,摇匀,0.45μm微孔滤膜滤过,作为供试品溶液。
2.2.4 阴性对照溶液 取处方组成中除大黄外的其他药材,制成不含大黄的阴性样品,按“2.2.3”项下方法处理制成阴性对照溶液。
2.2.5 运行缓冲液 精密称取硼砂3.81 g、磺丁基醚-β-CD 9.07 g、SDS 1.44 g,各置于100m l量瓶中,加水溶解并稀释至刻度,摇匀,作为贮备液。精密移取硼砂贮备液12.5m l、磺丁基醚-β-CD贮备液4m l、SDS贮备液20m l、甲醇1m l,置于50 m l量瓶中,加水稀释至刻度,摇匀,0.45μm微孔滤膜滤过,作为运行缓冲液。
2.3 专属性试验
分别吸取“2.2”项下对照品溶液(混合后加入内标贮备液)、供试品溶液、阴性对照溶液适量,按“2.1”项下电泳条件进样测定,记录色谱,详见图1。结果,在对照品色谱峰保留时间对应的位置上,供试品有色谱峰出现,且分离效果好,峰形对称,而阴性对照无干扰。
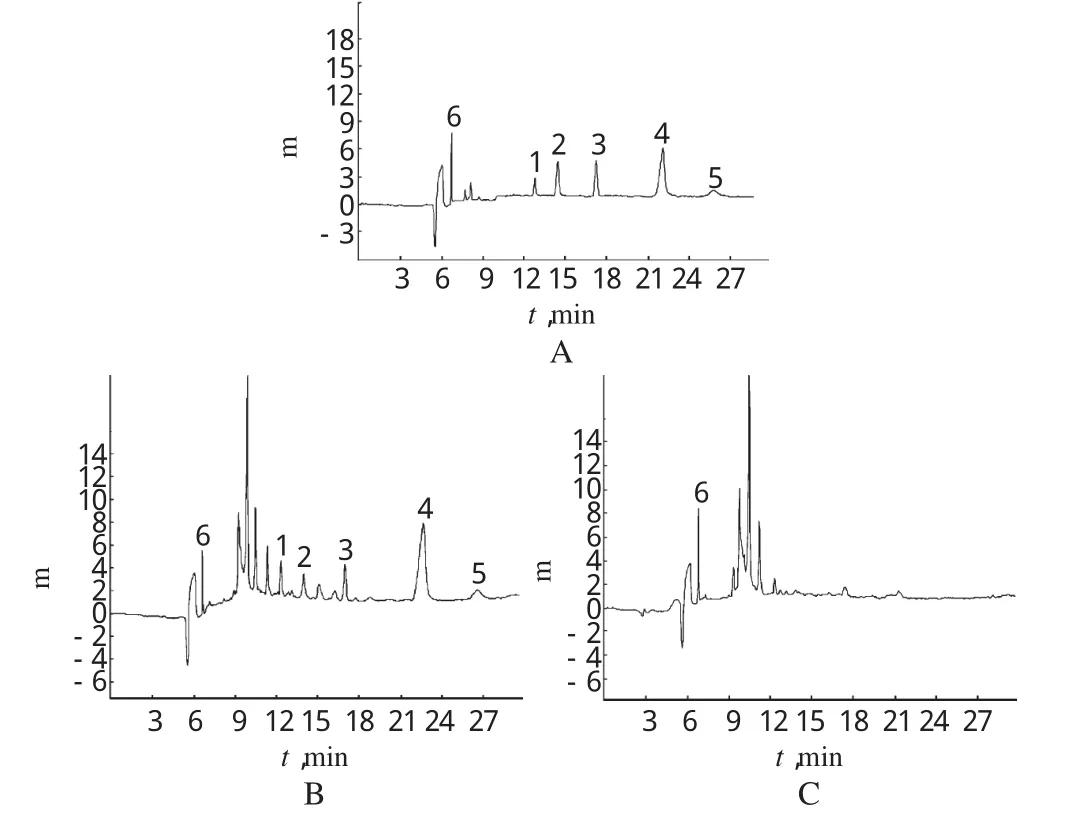
图1 毛细管电泳色谱图A.混合对照品+内标;B.供试品;C.阴性对照;1.大黄素;2.芦荟大黄素;3.大黄酸;4.大黄酚;5.大黄素甲醚;6.对乙酰氨基酚Fig 1 CE chromatogram sA.substance control+internal standard;B.test sample;C.negative control;1.emodin;2.aloe-emodin;3.rhein;4.chrysophanol;5.physcion;6.paracetamol
2.4 线性关系考察
分别吸取一定量的各对照品溶液,加甲醇稀释成大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚质量浓度分别为5.0、10.0、20.0、25.0、30.0、35.0、40.0μg/m l,2.4、4.8、9.6、12.1、14.4、16.8、19.6μg/m l,3.6、7.2、14.4、18.1、21.6、25.2、28.8μg/m l,11.2、22.5、45.0、56.2、67.4、78.7、89.9μg/m l,2.5、5.0、10.0、12.6、15.0、17.5、20.0μg/m l的系列溶液,加入适量内标贮备液使对乙酰氨基酚质量浓度为25μg/m l,分别进样。以各成分峰与内标峰面积的比值(y)为纵坐标,对照品溶液质量浓度(x)为横坐标,进行线性回归,分别得大黄素的回归方程y=0.045 4x+ 0.172 1(r=0.998 0)、芦荟大黄素的回归方程y=0.099 3x+ 0.019 5(r=0.997 1)、大黄酸的回归方程y=0.054 9x+0.060 6(r=0.996 9)、大黄酚的回归方程y=0.148 6x-0.293 3(r=0.998 5)、大黄素甲醚的回归方程y=0.092 0x+0.006 1(r=0.998 3)。结果表明,大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚检测质量浓度分别在5.0~40.0、2.4~19.2、3.6~28.8、11.2~89.9、2.5~20.0μg/m l范围内同各自峰面积与内标峰面积比值呈良好的线性关系。
2.5 精密度试验
制备质量浓度分别为25.0、12.1、18.1、56.2、12.6μg/m l的大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚对照品溶液,加入适量内标贮备液,分别进样,连续测定5次,记录峰面积并计算RSD。结果,5种成分峰面积与内标峰面积比值的RSD分别为1.08%、1.33%、2.40%、0.47%、1.05%,表明仪器精密度良好。
2.6 稳定性试验
取供试品溶液(批号:20100702)适量,在放置0、2、4、6、8、 10、12 h时分别进样,记录大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚的峰面积并计算RSD。结果,5种成分峰面积与内标峰面积比值的RSD分别为1.28%、2.13%、1.41%、0.99%、2.22%,表明供试品溶液在12 h内质量稳定。
2.7 重复性试验
取样品(批号:20100702)适量,共6份,按“2.2.3”项下方法制备供试品溶液,并按“2.1”项下电泳条件进样,测定并计算大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚的含量及RSD。结果,含5种成分的量平均分别为0.146、0.073、0.112、0.330、0.081mg/g,RSD分别为1.33%、0.72%、2.12%、2.26%、1.11%,表明本法重复性好。
2.8 加样回收率试验
精密称取同一批号样品(批号:20100702)适量,共6份,加入相应量的大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚对照品,按“2.2.3”项下方法处理并按“2.1”项下电泳条件进样测定,计算加样回收率及RSD,结果见表1。
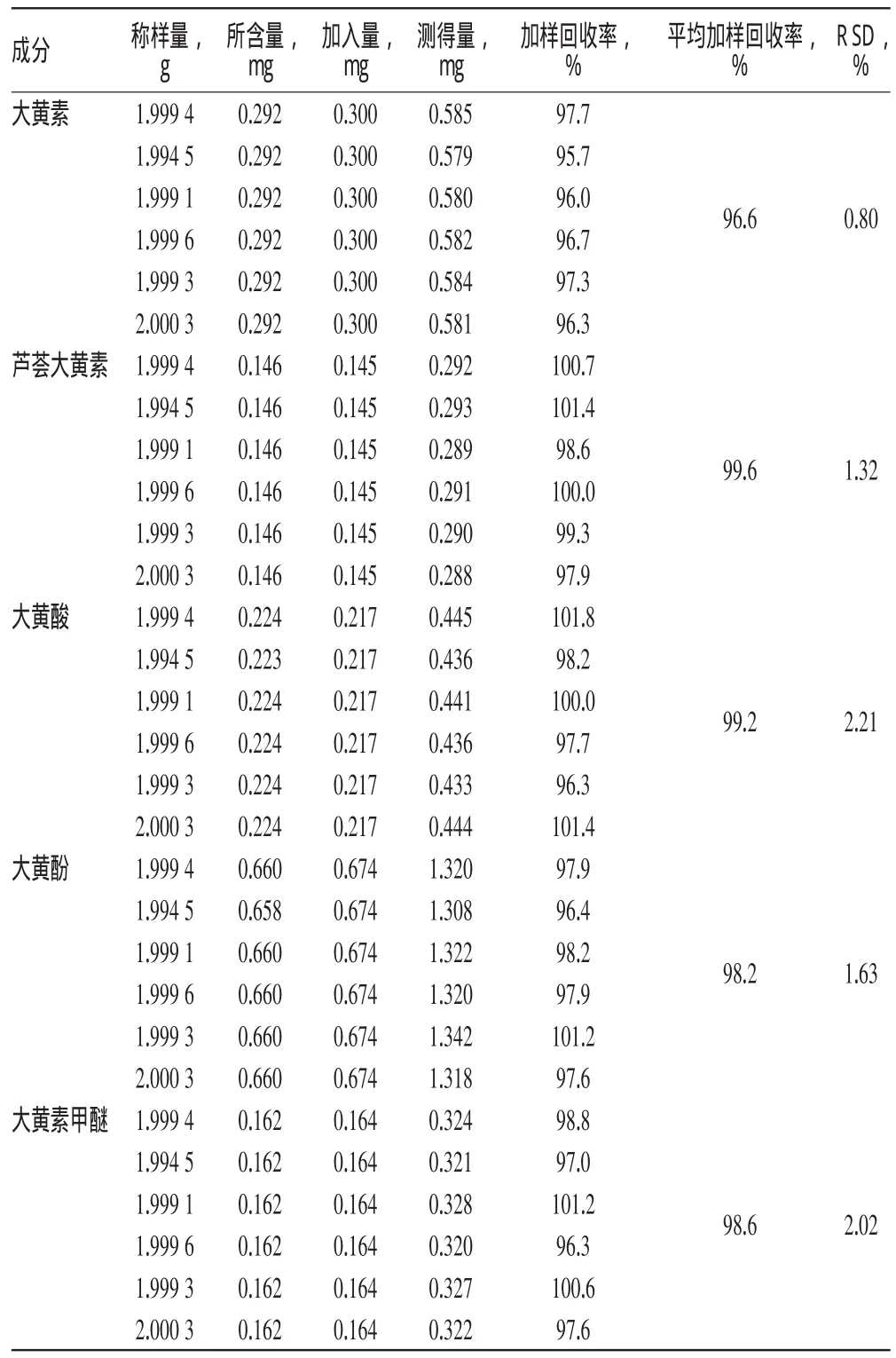
表1 加样回收率试验结果(n=6)Tab 1 Results of recovery tests(n=6)
2.9 样品含量测定
取样品3批,按“2.2.3”项下方法处理后,按“2.1”项下电泳条件进样测定,记录峰面积,代入回归方程计算其中大黄素、芦荟大黄素、大黄酸、大黄酚、大黄素甲醚的含量及RSD,结果见表2。
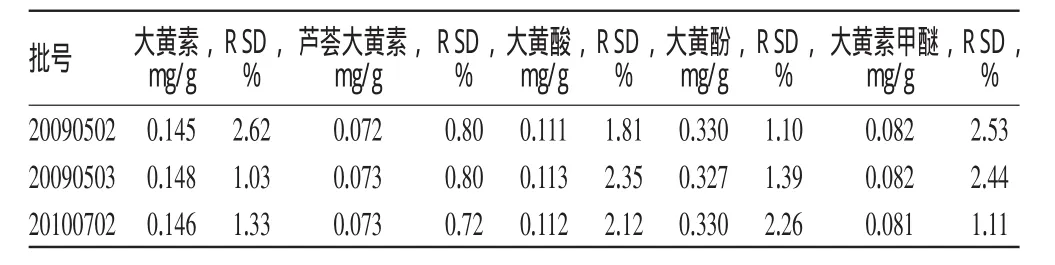
表2 样品含量测定结果(n=3)Tab 2 Results of content determ ination of sam p les(n=3)
3 讨论
3.1 供试品提取条件的选择
大黄中的蒽醌类成分大部分与糖结合,以蒽醌苷的形式存在于植物组织中,总量约3%~5%,少部分以游离形式存在。故可先用稀硫酸把蒽醌苷水解成苷元,此步骤采用超声提取可以提高效率和使水解更加充分完全,然后利用游离蒽醌可溶于热三氯甲烷的性质,用三氯甲烷加热回流将其提取出来,最后分取三氯甲烷层挥干后加乙醇超声进行结合蒽醌的提取。这样,就可以把大黄中的总蒽醌类成分全部提取出来。此外,本试验还考察了提取方法对样品中蒽醌类成分含量的影响。结果发现,单一的超声提取既无法完全提取总蒽醌,又有很多杂质影响目标峰,故选择超声提取法结合加热回流提取法。
3.2 检测波长的选择
大黄中重要的活性成分是蒽醌类化合物及其衍生物,根据化学结构可以推测其在230 nm、240~260 nm波长处有较大的吸收。本试验测定了5种成分在甲醇中的紫外吸收光谱,确定5种成分在254 nm波长处均有最大吸收,故选择254 nm为检测波长。
3.3 内标物的选择
为克服毛细管电泳法重复性差的缺点,本试验采用内标法定量。因所测物质为蒽醌类成分,蒽醌类化合物中多具有酚羟基,具有一定酸性,在缓冲体系中电离为阴离子,因此内标物也应选择能电离为阴离子的物质。笔者曾选择苯甲酸、对硝基苯甲酸、对氨基苯甲酸为内标物,但都因为与被测物质出峰时间太接近,达不到很好的分离效果。对乙酰氨基酚具有酚羟基,其结构与被测5种物质相近,在体系中能与其他物质分离,且无干扰,从而确定对乙酰氨基酚为本试验测定的内标物。
3.4 电泳条件的选择
3.4.1 硼砂浓度的影响 鉴于5种化合物均具有酚羟基,具有一定的弱酸性,为了这5种化合物更好地离子化,应选择碱性缓冲体系。本试验考察了磷酸盐、磷酸盐-硼酸、Tris、硼砂体系。发现磷酸盐体系电导率大,电流值也大,焦耳热大,难以形成平稳的基线。而Tris缓冲液的pH值受溶液浓度影响较大,且其在高浓度时的pH值不高,不能使待测物最大离子化,也易吸收空气中CO2,影响缓冲体系的质量。而硼砂缓冲液,其硼酸根能与多羟基化合物形成配位键,增加负电性,适用于其他含邻位羟基或多羟基化合物的分离,所以选择硼砂为缓冲试剂。参考文献[7-8]试验条件,在25mmol/L SDS+4mmol/L磺丁基醚-β-CD+8%甲醇基础上,本试验考察了硼砂溶液浓度分别为20、25、30mmol/L时对样品和对照品的分离情况。结果表明,硼砂溶液浓度越高,样品峰的拖尾因子越大,且一些目标峰的出现合并粘连现象,而浓度太低也会出现同样的情况,当硼砂溶液浓度为25mmol/L时的分离情况较好,故最后确定了此浓度。
3.4.2 SDS浓度的影响 根据5种化合物结构式可知,其结构相似,理化性质接近,故在区带模式下很难得到分离,此时需要考虑添加表面活性剂,常用SDS。当SDS浓度足够大,达到或超过临界浓度时,可形成胶束,而胶束在体系中的存在相当于固定相,溶质在胶束和水相之间进行分配,并由于其在胶束中不同的保留能力而产生差速迁移。在25mmol/L硼砂溶液+ 4mmol/L磺丁基醚-β-CD+8%甲醇条件下,本试验分别考察了15、20、25mmol/LSDS对样品分离情况的影响。结果发现,当SDS浓度为15mmol/L时,芦荟大黄素峰拖尾因子达不到要求,大黄素甲醚峰形不好,基线漂移(如图2A所示);当SDS浓度为25mmol/L时,各峰峰形较好,但大黄素甲醚峰无法达到基线分离(如图2 B所示);当SDS浓度为20mmol/L时,各目标峰峰形良好,且都能达到基线分离,在30min内完成分离测定(如图2 C所示)。故综合分离情况、分析时间等因素考虑,确定SDS浓度为20mmol/L。
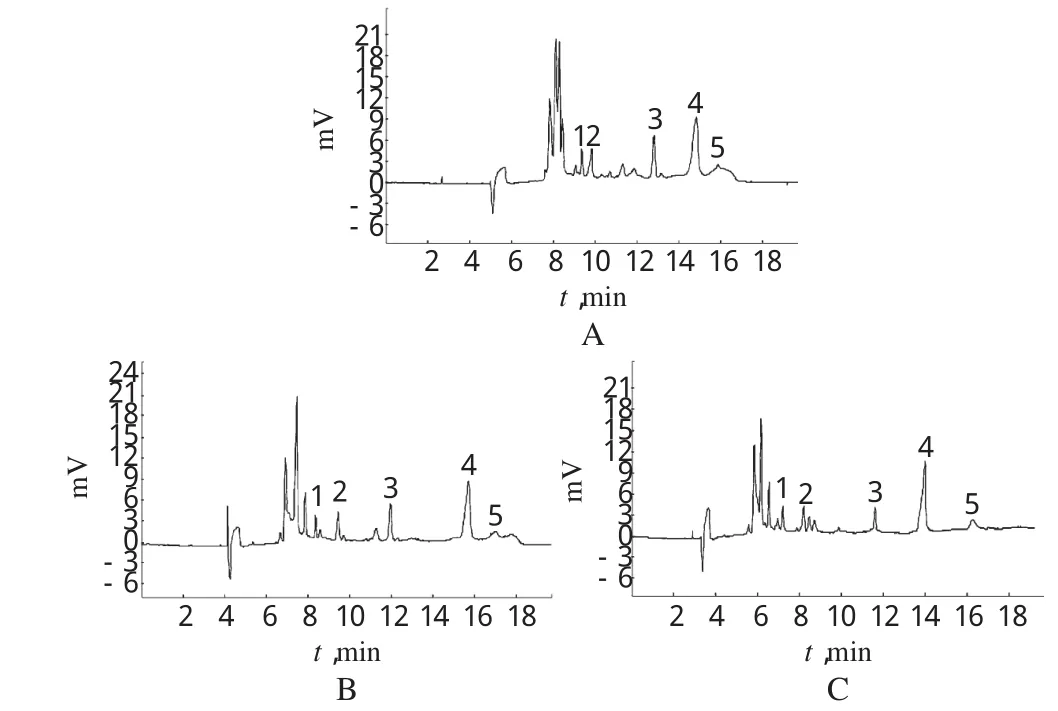
图2 5种成分在含不同SDS浓度的运行缓冲液中的电泳色谱图A.15mmol/L SDS+25mmol/L硼砂溶液+4mmol/L磺丁基醚-β-CD+ 8%甲醇;B.25 mmol/L SDS+25 mmol/L硼砂溶液+4 mmol/L磺丁基醚-β-CD+8%甲醇;C.20mmol/L SDS+25mmol/L硼砂溶液+4mmol/L磺丁基醚-β-CD+8%甲醇;1.大黄素;2.芦荟大黄素;3.大黄酸;4.大黄酚;5.大黄素甲醚Fig 2 Electrophorograms of 5 components in electrolyte w ith different concentrations of SDSA.15 mmol/L SDS+25 mmol/L Na2B4O7+4 mmol/L SBE-β-CD+8% methanol solution;B.25 mmol/L SDS+25 mmol/L Na2B4O7+4 mmol/L SBE-β-CD+8%methanol solution;C.20 mmol/L SDS+25 mmol/L Na2B4O7+4 mmol/L SBE-β-CD+8%methanol solution;1.emodin;2. aloe-emodin;3.rhein;4.chrysophanol;5.physcion
3.4.3 有机添加剂的选择 有机添加剂的加入能改变产生电流层的双电层,使电渗流降低,扩大迁移窗口,增加峰容量,提高分离效率。目前用的最多的是甲醇和乙腈两种有机溶剂,就降低电渗而言甲醇等醇类的作用比乙腈更大。本试验选择甲醇为有机添加剂,并考察了不同比例(2%、5%、8%)甲醇对分离情况的影响。结果,综合分离效率和迁移时间,最终选择甲醇添加比例为2%。
3.4.4 磺丁基醚-β-CD的影响 CD具有中内疏水、外亲水的圆筒结构,能与多种疏水性的化合物形成包合物,其不仅是手性选择剂,而且被广泛用作胶束电动毛细管色谱(MECC)法的添加剂。CD在分离中的作用之一是立体空间的选择性。较小的溶质分子可以进入CD疏水性空腔内部,较大的分子则受到限制,从而产生对溶质分子大小的选择性[9]。本试验发现,加入磺丁基醚-β-CD后,峰形变窄,分离效果好。进而考察了磺丁基醚-β-CD浓度对分离情况的影响。结果发现,当其浓度过大时,可能由于其包合作用使得本来响应值不高的大黄素甲醚峰消失;当其浓度太小时,样品目标峰会受其他杂质峰干扰。综合考虑对分离和迁移时间的影响,本试验最终选择磺丁基醚-β-CD的浓度为4mmol/L。
3.4.5 pH的影响 在上述条件确定的基础上,本试验考察了不同pH对峰分离的影响。结果发现,pH小于10.00时(9.54)大黄素甲醚峰无法分离,而pH大于10.00时(10.32)大黄素甲醚峰达到分离效果,但是大黄酚峰拖尾严重。综合考虑,确定pH为10.01。
3.4.6 分离电压的影响 分离电压升高,分离时间变短,产生的焦耳热增多,会引起区带展宽,谱峰的基线过高且部分峰重叠;分离电压过低,则分离时间较长,且分离不完全。在缓冲体系确定为25mmol/L硼砂溶液+20mmol/L SDS+4mmol/L磺丁基醚-β-CD+2%甲醇(pH 10.01)时,本试验考察了在15~25 kV范围内的最适分离电压。综合基线平滑度、峰展宽、柱效、分离度、保留时间、拖尾因子等考虑,最后确定分离电压为18 kV。
[1]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:1 116-1 117.
[2]卫恒巧,李作平,崔秀彦,等.薄层扫描法测定清肺抑火丸中小檗碱和菝葜皂苷元的含量[J].河北医科大学学报,1992,13(4):199.
[3]黄瑞红,杨慧文.RP-HPLC法测定清肺抑火丸中白花前胡甲素的含量[J].中国药房,2010,21(27):2 556.
[4]叶秀金,宋粉云.HPLC测定清肺抑火丸中盐酸小檗碱的含量[J].中国实验方剂学杂志,2011,17(3):90.
[5]叶秀金,宋粉云.HPLC法测定清肺抑火丸中大黄的四种成分的含量[J].中药新药与临床药理,2010,21(6):646.
[6]陈峻,徐华,陈小轩.HPLC同时测定清肺抑火丸中3个苷类成分[J].中国实验方剂学杂志,2013,19(13):134.
[7] 刘丹,毋福海,曾承辉.毛细管电泳法测定一清颗粒中七种成分的含量[J].中国医药工业杂志,2009,40(11):840.
[8] 李艺,朱培仪,宋粉云.毛细管电泳分离与测定大鼠血浆中尼群地平和尼莫地平对映体的浓度[J].药物分析杂志,2011,31(2):306.
[9] 徐其进,顾中伟,陈先丽.疏水条件下甾体化合物的分离及环糊精的作用[J].分析化学,1999,27(2):193.
Simultaneous Determ ination of 5 Anthraquinones in QingfeiYihuo Pills by Capillary Electrophoresis
CHEN Cen,WANG Xiao-ke,WU Fu-hai(Guangdong Pharmaceutical University,Guangzhou 510224,China)
OBJECTIVE:To establish a method for simultaneous determ ination of 5 anthraquinones in Qingfei yihuo pills,such as emodin,aloe-emodin,rhein,chrysophanol and physcion.METHODS:Capillary electrophoresis was adopted using paracetamol as internal standard.The separation was performed on a uncoated fused silica capillary.25 mmol/L Na2B4O7+20mmol/L SDS +4 mmol/L SBE-β-CD+2%methanol solution(pH 10.01)was selected as the running buffer.The voltage applied was 18 kV.The sample was injected by gravity(10 s,15 cm).The detection wavelength was set at 254 nm.The column temperature was 25℃and hum idity was lower than 70%.RESULTS:The linear range of emodin,aloe-emodin,rhein,chrysophanol,physcion were 5.0-40.0 μg/m l(r=0.998 0),2.4-19.2μg/m l(r=0.997 1),3.6-28.8μg/m l(r=0.996 9),11.2-89.9μg/m l(r=0.998 5),2.5-20.0μg/m l(r=0.998 3),respectively;RSDs of precision,stability and reproducibility tests were all lower than 2.40%;average recoveries were 96.6%(RSD=0.80%,n=6),99.6%(RSD=1.32%,n=6),99.2%(RSD=2.21%,n=6),98.2%(RSD=1.63%,n=6)and 98.6%(RSD=2.02%,n=6).CONCLUSIONS:The method is proved to be specific,accurate,reliable and reproducible,and can be used for quality control of Qingfei yihuo pills.
Capillary electrophoresis;Qingfei yihuo pills;Emodin;A loe-emodin;Rhein;Chrysophanol;Physcion;Content determ ination
R917
A
1001-0408(2014)12-1118-04
DOI 10.6039/j.issn.1001-0408.2014.12.22
广东省自然科学基金资助项目(No.5002841)
2013-10-29
2014-01-26)
