成纤维细胞生长因子及其受体抑制剂的研究进展
2014-01-14杜培娟
杜培娟
(青岛科技大学化工学院,山东 青岛266042)
成纤维细胞生长因子受体(FGFRs)是受体酪氨酸激酶(RTKs)超家族的一员,它具有胞外配体结构域和胞内酪氨酸激酶结构域。RTKs是一大类酶联受体,有50多种类型,主要类型有表皮生长因子受体(EGFR)家族、胰岛素受体(IR)家族、血小板衍生生长因子受体(PDGFR)家族、血管内皮生长因子受体(VEGFR)家族、FGFRs家族、肝细胞生长因子受体(HGF)家族、神经生长因子受体(NGF)家族[1]等。RTKs参与胚胎及成年组织中各种生理反应的动态平衡。在癌变过程中,由于RTKs异常或致癌基因激活而导致肿瘤细胞增生和抗凋亡,这一过程称为RTKs致癌成瘾,简称为致癌成瘾[2]。由于具有致癌成瘾的癌细胞对相应的RTKs抑制剂非常敏感,所以RTKs是最受关注的抗癌药物靶标[3]。FGFRs作为RTKs超家族的一员,已成为全球制药公司开发新型抗肿瘤药物的靶标之一。
1 成纤维细胞生长因子(FGFs)及其受体(FGFRs)
1.1 FGFs
FGFs是一类由FGF基因编码的结构相关的蛋白质,其分子量一般为20~30ku[4]。FGFs家族包括23种配体,其中分泌型配体18种,分别为FGF1(aFGF)、FGF2(bFGF)、FGF3(INT2)、FGF4、FGF5、FGF6、FGF7(KGF)、FGF8、FGF9、FGF10、FGF16、FGF17、FGF18、FGF19、FGF20、FGF21、FGF22和FGF23[5]。FGFs能够结合多种成纤维细胞生长因子受体(FGFRs),并与硫酸乙酰肝素蛋白多糖(HSPGs)、Klotho家族成员等以组织特异性方式相互协调,从而促进FGFs信号的传导[6]。
1.2 FGFRs
FGFRs家族包括以下类型:FGFR1b、FGFR1c、FGFR2b、FGFR2c、FGFR3b、FGFR3c、FGFR4。它们具有共同的结构域,包括胞外免疫球蛋白样结构域和胞内酪氨酸激酶结构域[5]。FGFR1的基因在人类染色体的8p12位点编码FGFR1b和FGFR1c亚型,由于选择性剪接作用,它们在第三免疫球蛋白样结构域存在差异。FGFR2基因在人类染色体的10q26位、FGFR3基因在人类染色体的4p16.3位也是编码两种型式。在癌细胞中,人类原癌基因由于基因扩增、染色体易位和点突变等而发生活化产生FGFRs基因。FGFRs分别在癌细胞和内皮细胞中参与肿瘤的发生和血管的生成,因此,FGFRs-靶向药物会产生直接或间接的抗癌作用[7]。
2 FGFs信号的激活和传导
FGFs能够在酪氨酸激酶结构域中的关键活化环的酪氨酸残基上引发FGFRs的自身磷酸化,从而导致酪氨酸激酶结构域从非活化状态转变为活化状态[8]。FGFRs中活化的酪氨酸激酶结构域在底物结合位点沿着FGFRs结合的衔接分子逐步磷酸化其它酪氨酸残基。FGFRs的C-末端区的酪氨酸残基磷酸化能够使磷酸酯酶Cγ(PLCγ)吸纳并激活,从而催化磷脂酰肌醇二磷酸(PIP2)转化为甘油二脂(DAG)和三磷酸肌醇(IP3)[9]。活化的FGFR磷酸化底物2(FRS2)能够吸纳生长因子受体结合蛋白2(GRB2)适配分子。
FGFs信号可以通过FRS2和GRB2传导到Ras-促分裂原活化蛋白激酶(Ras-MAPK)或PI3激酶-蛋白激酶B(PI3K-AKT)信号通路,通过PLCγ和DAG传导到蛋白激酶C(PKC)或蛋白激酶D(PKD)信号通路,通过PLCγ和IP3传导到钙离子释放级联通路。FGFs诱导的Ras-MAPK活化参与细胞增殖,而FGFs诱导的PI3K-AKT活化参与细胞存活。Rab5小分子鸟苷三磷酸酶是一个活化的FGFRs结合分子,它能够参与Ras-MAPK信号而非PI3K-AKT信号的持续传导[10]。FGFs信号引发的靶细胞的各种反应见图1[11]。
3 FGFR抑制剂
由于FGFs信号参与肿瘤生物学的各个方面,如抗凋亡、血管生成、EMT和侵袭等(图1),FGFRs的靶向治疗已成为临床肿瘤学领域的热点,拟合于酪氨酸激酶结构域中ATP结合口袋而设计开发的小分子化合物已经用于癌症治疗。
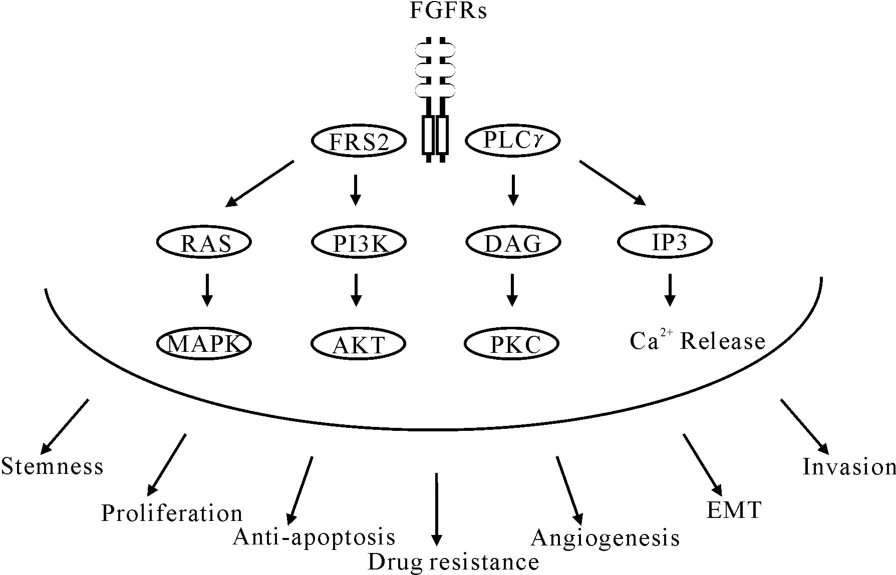
图1 FGFs信号及其引发的靶细胞的各种反应Fig.1 FGFs Signaling and the responses of target cells
ponatinib(AP24534)、PD173074、MK-2461、brivanib alaninate、danusertib(PHA739358)、nintedanib(BIBF1120)、BGJ398(NVP-BGJ398)、dovitinib(TKI258)、AZD4547和SSR128129E是靶向FGFRs的酪氨酸激酶抑制剂(TKIs),即FGFR抑制剂,其结构式如图2所示。它们具有良好的FGFRs抑制活性,酶抑制活性的IC50基本上都在纳摩尔水平(表1)。
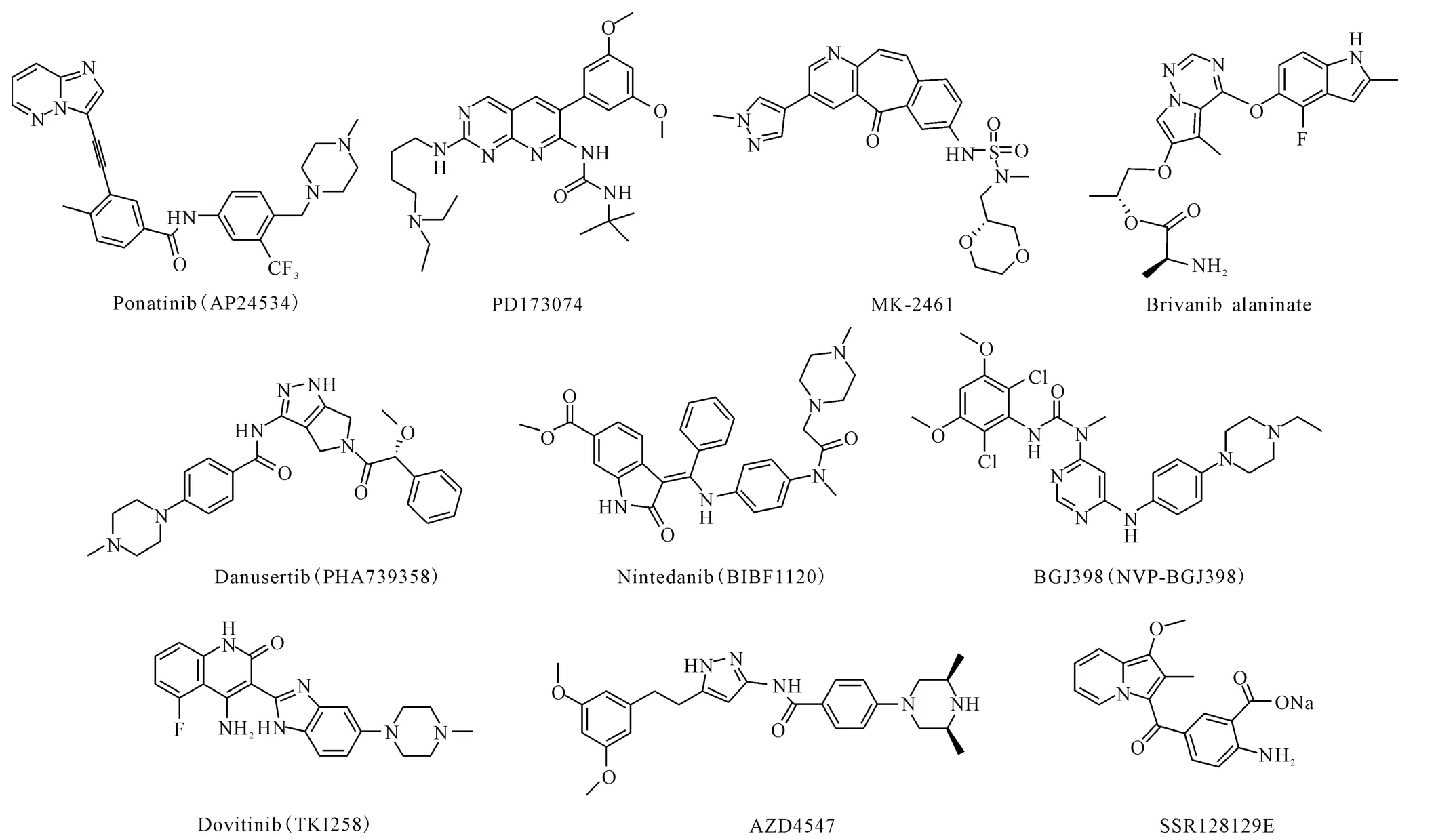
图2 FGFR抑制剂的结构式Fig.2 Structures of FGFR inhibitors

表1 FGFR抑制剂的酶抑制活性IC50/(nmol·L-1)Tab.1 Enzyme inhibitory activity IC50of FGFR inhibitors/(nmol·L-1)
3.1 第一代FGFR抑制剂
ponatinib(AP24534)、dovitinib(TKI258)、PD173074、danusertib(PHA739358)、MK-2461、nintedanib(BIBF1120)和brivanib alaninate(BMS582664)是靶向FGFRs和其它RTKs的第一代FGFR抑制剂。
ponatinib(AP24534)[12]是口服有效的多靶点激酶抑制剂,它能够有效地抑制FGFR1(IC50为2nmol·L-1),还可以抑制PDGFRα、FLT3和c-KIT,IC50分别为1nmol·L-1、13nmol·L-1、13nmol·L-1。由于ponatinib(AP24534)作用于表达野生型或T315I突变型BCR-ABL的CML细胞系,能够有效抑制BCR-ABL调节的信号,因此ponatinib(AP24534)在Ⅱ期临床试验阶段作为BCR-ABL抑制剂使用。目前,ponatinib(AP24534)对非小细胞肺癌的治疗也进入了Ⅱ期临床试验阶段。
dovitinib(TKI258)[13-14]是口服生物利用度好 的FGFR抑制剂,不仅能够有效抑制FGFR1和FGFR2(IC50分别为8nmol·L-1和9nmol·L-1),还能够抑制VEGFR2、FLT3、c-KIT等靶点。dovitinib(TKI258)能够抑制由FGFRs活化异常而导致的癌细胞的体外增殖,并且在临床前异种移植模型试验中诱导肿瘤消退。目前,dovitinib(TKI258)对肾癌的治疗进入了Ⅲ期临床试验,对晚期乳腺癌、子宫内膜癌、复发性多发性骨髓瘤和膀胱上皮癌的治疗进入了Ⅱ期临床试验。
PD173074[15-16]是FGFR1的ATP竞争性抑制剂,IC50约为25nmol·L-1,也能抑制VEGFR2,IC50为100~200nmol·L-1,作用于FGFR1比作用于PDG-FR和c-Src选择性高1 000倍左右。它不仅能够以剂量依赖的方式抑制FGFR1和FGFR3的磷酸化(IC50为1~5nmol·L-1),还能够特异性地抑制FGF2介导的少突胶质细胞(OL)谱系细胞的增殖、分化和MAPK激活。
danusertib(PHA739358)[17]是一种aurora kinase抑制剂,作用于aurora A/B/C时IC50为13nmol·L-1/79nmol·L-1/61nmol·L-1,研究发现其对FGFR1也有抑制作用,IC50为47nmol·L-1。danusertib(PHA739358)对难治性前列腺癌及多发性骨髓瘤的治疗进入了Ⅱ期临床试验阶段。
MK-2461[18]是一种有效的多靶点抑制剂,能有效抑制FGFR1、FGFR2、FGFR3、c-Met、Ron和FLT3,IC50分别为65nmol·L-1、39nmol·L-1、50nmol·L-1、2.5nmol·L-1、7nmol·L-1和10nmol·L-1。MK-2461明显抑制广谱肿瘤细胞系增殖,特别是针对高表达MET和FGFR2的癌细胞。
nintedanib(BIBF1120)[19]是一种有效的三重血管激酶抑制剂,对FGFR1/2/3、VEGFR1/2/3和PDGFRα/β均有抑制作用,其IC50分别为69nmol·L-1/37nmol·L-1/108nmol·L-1、34nmol·L-1/13nmol·L-1/13nmol·L-1和59nmol·L-1/65 nmol·L-1。nintedanib(BIBF1120)已用于治疗非小细胞肺癌、前列腺癌、卵巢癌及结肠直肠癌等。目前,其对宫颈癌、间皮瘤以及非小细胞肺癌等的治疗已进入了Ⅱ期临床试验阶段,对特发性肺纤维化的治疗进入了Ⅲ期临床试验阶段。
brivanib alaninate(BMS582664)[20]是BMS540215的丙氨酸前体药物,是一种ATP竞争性的RTKs抑制剂。它能够抑制FGFR1和VEGFR2的磷酸化,阻断FGF和VEGF的信号传导,从而抑制肿瘤生长。
3.2 第二代FGFR抑制剂
AZD4547、BGJ398(NVP-BGJ398)和SSR128129E是针对FGFR开发的第二代抑制剂,其对FGFR具有良好的选择性。
AZD4547[21]是一种新型的选择性FGFR抑制剂,靶向作用于FGFR1、FGFR2和FGFR3,IC50分别为0.2nmol·L-1、2.5nmol·L-1和1.8nmol·L-1,对FGFR4、VEGFR2(KDR)的抑制活性较弱,对IGFR、CDK2和p38几乎没有抑制活性。目前,AZD4547对乳腺癌及非小细胞肺癌的治疗进入了Ⅱ期临床试验阶段,对实体瘤的治疗进入了Ⅰ期临床试验阶段。
BGJ398(NVP-BGJ398)[22]是高选择性的FGFR抑制剂,作用于FGFR1、FGFR2和FGFR3,IC50分别为0.9nmol·L-1、1.4nmol·L-1、1nmol·L-1,对Abl、Fyn、Kit等靶点几乎没有抑制活性。BGJ398(NVP-BGJ398)不仅能够抑制FGFR1、FGFR2和FGFR3依赖的BaF3的细胞增殖,还抑制过量表达野生型FGFR3的癌细胞的增殖,其对复发性胶质母细胞瘤及黑色素瘤的治疗进入了Ⅱ期临床试验阶段。
SSR128129E[23]是口服效果较好、选择性较高的FGFR抑制剂,其抑制FGFR1的IC50为1.9μmol·L-1,对其它的关联RTKs无效果。在患有关节炎的小鼠身上,SSR128129E能够抑制血管生成、炎症和骨吸收,并且缓解了临床症状。在各种小鼠肿瘤模型中,SSR128129E同时抑制了原发性肿瘤的增殖和转移。
4 结语
在基于FGFRs异常激活而获得抗凋亡潜能的人类癌细胞中,抑制FGFs信号能够在抑制血管新生的同时降低癌细胞的负荷,并且FGFR抑制剂能够增强癌细胞对常规抗癌药物(如5-氟尿嘧啶、伊立替康、紫杉醇等)的敏感性。随着人们对FGFs信号网络的深入了解,以及对FGFs与FGFRs作用机制的深入研究,特异性强、治疗效果好的FGFR抑制剂将会被开发出来。采用FGFRs-靶向抗癌药物治疗肿瘤将会具有非常广阔的前景。
[1]曹川,戴霞,李世荣.生长因子受体酪氨酸激酶的结构与功能[J].现代生物医学进展,2008,8(5):952-954.
[2]WEINSTEIN I B,JOE A K.Mechanisms of disease:Oncogene addiction——a rationale for molecular targeting in cancer therapy[J].Nature Clinical Practice Oncology,2006,3(8):448-457.
[3]ZHANG J M,YANG P L,GRAY N S.Targeting cancer with small molecule kinase inhibitors[J].Nature Reviews Cancer,2009,9(1):28-39.
[4]许英蕾,孙建义.成纤维细胞生长因子与其受体的研究进展[J].药物生物技术,2004,11(3):194-198.
[5]ESWARAKUMAR V P,LAX I,SCHLESSINGER J.Cellular signaling by fibroblast growth factor receptors[J].Cytokine & Growth Factor Reviews,2005,16(2):139-149.
[6]MASOLA V,GAMBARO G,TIBALDI E,et al.Heparanase and syndecan-1interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells[J].Journal of Biological Chemistry,2012,287(2):1478-1488.
[7]GREULICH H,POLLOCK P M.Targeting mutant fibroblast growth factor receptors in cancer[J].Trends in Molecular Medicine,2011,17(5):283-292.
[8]BAE J H,SCHLESSINGER J.Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases[J].Molecules and Cells,2010,29(5):443-448.
[9]DAILEY L,AMBROSETTI D,MANSUKHANI A,et al.Mechanisms underlying differential responses to FGF signaling[J].Cytokine & Growth Factor Reviews,2005,16(2):233-247.
[10]VECCHIONE A,COOPER H J,TRIM K J,et al.Protein partners in the life history of activated fibroblast growth factor receptors[J].Proteomics,2007,7(24):4565-4578.
[11]KATOH M,NAKAGAMA H.FGF Receptors:Cancer biology and therapeutics[J].Medicinal Research Reviews,2014,34(2):280-300.
[12]GOZGIT J M,WONG M J,MORAN L,et al.Ponatinib(AP24534),a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models[J].Molecular Cancer Therapeutics,2012,11(3):690-699.
[13]TRUDEL S,LI Z H,WEI E,et al.CHIR-258,a novel,multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14)multiple myeloma[J].Blood,2005,105(7):2941-2948.
[14]CHASE A,GRAND F H,CROSS N C P.Activity of TKI258against primary cells and cell lines with FGFR1fusion genes associated with the 8p11myeloproliferative syndrome[J].Blood,2007,110(10):3729-3734.
[15]DIMITROFF C J,KLOHS W,SHARMA A,et al.Anti-angiogenic activity of selected receptor tyrosine kinase inhibitors,PD166285and PD173074:Implications for combination treatment with photodynamic therapy[J].Investigational New Drugs,1999,17(2):121-135.
[16]BANSAL R,MAGGE S,WINKLER S.Specific inhibitor of FGF receptor signaling:FGF-2-mediated effects on proliferation,differentiation,and MAPK activation are inhibited by PD173074in oligodendrocyte-lineage cells[J].Journal of Neuroscience Research,2003,74(4):486-493.
[17]CARPINELLI P,CERUTI R,GIORGINI M L,et al.PHA-739358,apotent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer[J].Molecular Cancer Therapeutics,2007,6(12):3158-3168.
[18]PAN B S,CHAN G K Y,CHENARD M,et al.MK-2461,a novel multitargeted kinase inhibitor,preferentially inhibits the activated c-Met receptor[J].Cancer Research,2010,70(4):1524-1533.
[19]HILBERG F,ROTH G J,KRSSAK M,et al.BIBF 1120:Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy[J].Cancer Research,2008,68(12):4774-4782.
[20]HUYNH H,NGO V C,FARGNOLI J,et al.Brivanib alaninate,a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases,induces growth inhibition in mouse models of human hepatocellular carcinoma[J].Clinical Cancer Research,2008,14(19):6146-6153.
[21]GAVINE P R,MOONEY L,KILGOUR E,et al.AZD4547:An orally bioavailable,potent,and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family[J].Cancer Research,2012,72(8):2045-2056.
[22]GUAGNANO V,FURET P,SPANKA C,et al.Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea(NVPBGJ398),apotent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase[J].Journal of Medicinal Chemistry,2011,54(20):7066-7083.
[23]BONO F,de SMET F,HERBERT C,et al.Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties[J].Cancer Cell,2013,23(4):477-488.
