基于matK基因序列的核桃种间亲缘关系分析
2014-01-14宋艳波梅霞王勇王金胜吴国良
宋艳波,梅霞,王勇,王金胜,吴国良
(1.山西农业大学生命科学学院,山西太谷 030801;2.山西省农业科学院果树研究所,山西太谷 030801;3.河南农业大学园艺学院,郑州 450002)
基于matK基因序列的核桃种间亲缘关系分析
宋艳波1,梅霞1,王勇2,王金胜1,吴国良3*
(1.山西农业大学生命科学学院,山西太谷 030801;2.山西省农业科学院果树研究所,山西太谷 030801;3.河南农业大学园艺学院,郑州 450002)
matK基因是陆地植物核心条码候选片段之一,文章通过克隆获得22个核桃品种matK基因近3′端长度为726 bp EST序列,运用序列分析技术探索matK在核桃种间关系研究中的价值。结果表明,在所测的matK基因的核苷酸序列中发生变异的核桃品种均属早实核桃类群,而晚实核桃类群无变异发生;推导氨基酸序列分析显示薄壳香由于发生突变,提前终止翻译而只编码173个氨基酸,而其他品种均编码234个氨基酸;采用近邻接树法和最大简约法构建的系统进化树显示,22个品种核桃分为5个组,其中扎343与其他21个品种有明显差异,8个新选育的品种中金薄丰1号较为突出;而辽核1号与西扶1号亲缘关系最近。对matK基因深入研究将为核桃种质资源保护、杂交育种及开发运用提供分子生物学依据。
核桃;matK序列;早实类群;进化树
核桃(Juglans regia L.)隶属胡桃科(Juglandace⁃ae)核桃属(Juglans),种质资源丰富,共有13个种,分布于中国24个省区,栽培面积和产量均居世界首位[1]。但我国核桃单位面积产量极低,不到美国1/10,原因之一是良种化程度低[2]。近年来注重核桃早实品种选育,出现大批核桃新品种和品系,但品种区分易出现混乱[3],不利于核桃资源保护、保存及开发利用。对核桃种质资源的研究主要集中于两个方面:利用RAPD等分子标记技术对种质资源展开遗传多样性分析[4-5];分析早晚实核桃品种特性[6-7]。虽在种质资源鉴定和品种间亲缘关系方面取得进展,但分子标记技术需对多对引物进行筛选,存在工作量大、需时长、耗资大等缺点,因此寻找快速高效的核桃资源鉴定和品种遗传多样性研究方法仍是目前亟待解决难题。
Paul Hebert提出DNA条形码技术。DNA条形码(DNA barcode)是生物体内能够代表该物种的、标准的、有一定变异程度的、易扩增且相对较短的DNA片段[8],是一项新兴技术,由于其准确性高、操作简便且快速,已成为物种鉴定以及发现新的或隐性物种的有用工具。2009年国际条形码联盟将matK基因推荐为陆地植物核心条码候选片段之一[9],该基因位于叶绿体基因组上的赖氨酸trnK基因的内含子内,可能参与RNA转录体中II型内含子剪切,是叶绿体蛋白编码基因中进化速率最快的单编码基因之一。研究表明,对matK基因进行序列分析可为属及属以下水平的种群内部系统重建研究提供较多信息和较高支持率[10]。该基因序列碱基转换和颠换频率以及密码子位置变异概率都不高。因此,在构建系统树时,不需对不同类型的变异进行加权,保证分子系统树可靠性。
近年来,运用matK基因序列进行多种植物的系统发育及鉴定研究较多[11],而被子植物matK基因序列变异较大,对构建比较古老物种的科内系统树更有利[12]。该法在中药材等植物的鉴别和发育研究中被广泛应用[13]。但木本类植物运用此技术研究较少,且对种属间关系展开探讨。
本文利用matK序列分析技术,对山西省农业科学院果树研究所22个栽培核桃品种(其中包括最新实生选育的8个品种)构建NJ系统进化树和MP系统进化树,探究核桃系统发育关系,从分子水平研究核桃种内遗传多样性提供技术支持,为核桃品种鉴定和种质资源筛选、保存及利用提供科学依据。
1 材料与方法
1.1 材料
2011年7月在山西省农业科学院果树研究所采摘如下22个核桃品种的叶片:薄壳香,香铃,扎343,981,京861,西林3号,辽核1号,中林1号,西扶1号,礼品1号,礼品2号,清香,晋龙1号,晋龙2号,以及山西省最新实生选育的金薄丰1号和金薄香系列(金薄香1号、2号、3号、4号、6号、7号和8号)。
1.2 总基因组DNA的提取、PCR扩增及序列测定
核桃叶片DNA的提取参照宋艳波等改良CTAB法进行[14]。
搜索GenBank数据库中核桃科植物matK基因的EST数据进行体外拼接,进而根据拼接序列设计matK基因的特异引物对5′TCCTTTCATTCATTAT⁃GTTAGA 3′/5′GTTGTTGGTCTCATAATCAAT 3′。以不同品种核桃的基因组DNA为模板,进行PCR扩增获得目的基因片段,扩增程序为94℃预变性3 min;94℃变性40 s,37.2℃退火30 s,72℃延伸60 s,35个循环;72℃延伸10 min。将回收纯化的目的片段与P-BLUE-T(CV04,Aidlab)载体进行连接并转化大肠杆菌(E.coli)DH5a感受态细胞,通过蓝白斑选取阳性克隆子,扩大培养,菌液PCR反应检测后,委托北京英骏公司进行测序反应,每反应测序2次。
1.3 不同核桃品种matK基因片段的序列分析
对测定的22个序列手动去除载体后,使用NCBI中BLAST数据库进行序列比对,确定克隆结果。再利用DNASTAR软件中MegAlight模块进行多重序列比对,分析品种间DNA差异位点并生成遗传距离图谱。
1.4 核桃种间和属间进化分析及系统进化树建立
利用DNASTAR软件中SeqBuider模块将所得22个核桃品种matK基因片段序列翻译成相应的氨基酸序列,并对所得的氨基酸序列进行多重比对。之后,利用Mega 5.0软件构建系统进化树,建树方法采用最大邻接法(Neighbor-Joining,NJ)及最大简约法(Maxium parsimony,MP)。树的准确性用1 000次bootstrap值加以判断。
2 结果与分析
2.1 核桃matK基因片段的克隆
以不同品种核桃的基因组DNA为模板,经过PCR扩增均获得1条约750 bp特异片段(见图1),序列比较分析显示供试品种该基因片段序列长度均为726 bp,部分序列已提交到GenBank,登录号为KC533799、KC533800、KC920668-KC920677。
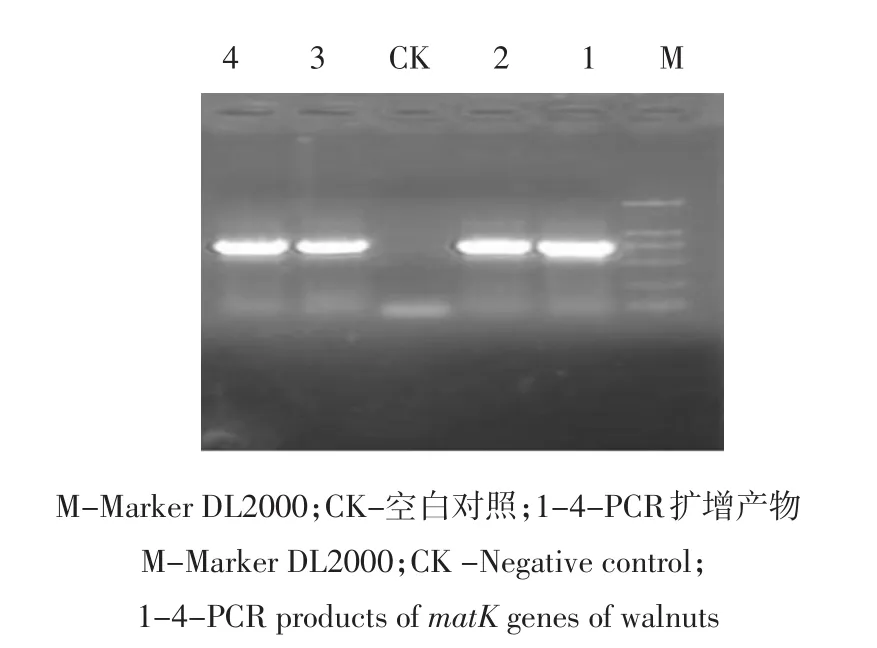
图1 核桃matK基因片段的PCR扩增Fig.1 PCR product of matK gene in walnut
2.2 不同品种核桃matK基因片段核酸序列分析
应用DNAStar软件对22个核桃品种matK基因片段的核酸序列进行聚类比较分析,其差异位点如表1所示。
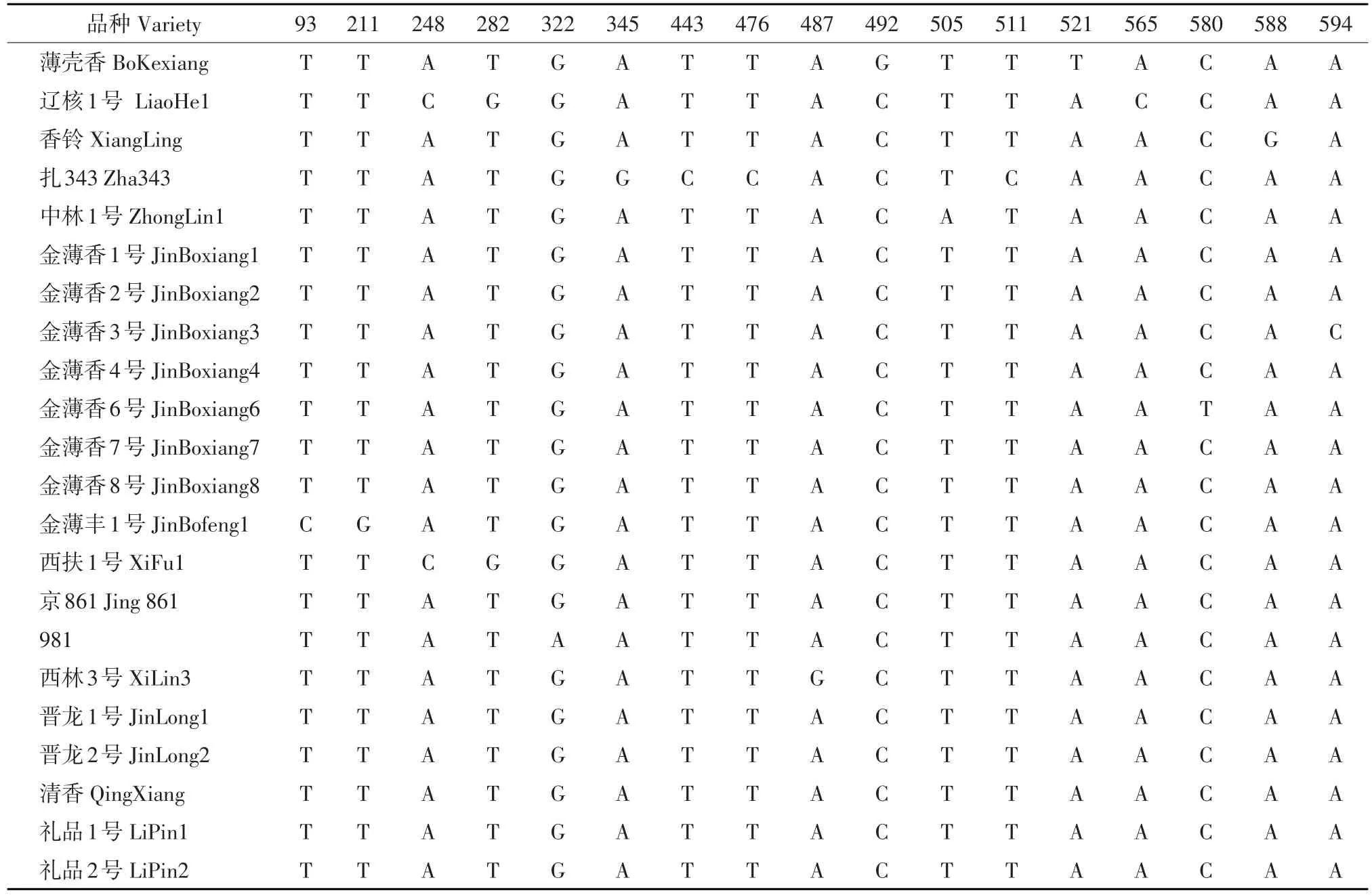
表1 22个核桃品种matK序列差异位点Table 1 Analysis of differences sites between 22 walnut matK sequence
22个核桃品种中发现有17个SNP位点,分别出现在93、443、476和511号位置的T-C转换,322号位置的G-A转换,345、487和588号位置的A-G转换,580号位置的C-T转换;211和282号位置的T-G颠换,248、565和594号位置的A-C颠换,492号位置的C-G颠换,505和521号位置的A-T颠换。其中,发生转换频率为47.1%,发生颠换频率为52.9%。这与遗传学上颠换较转换频率高不符,可能与片段较小有关,不能代表物种整体的转换颠换频率。但从表中可见,扎343有4个变异位点,辽核1号有3个变异位点,薄壳香、金薄丰1号、西扶1号有2个变异位点,金薄香3号、金薄香6号、中林1号、981及香玲有1个变异位点,这些存在变异位点的核桃品种均属早实核桃类群;而礼品1号、礼品2号、晋龙1号及晋龙2号属典型的晚实核桃,序列完全相同。由于matK基因参与RNAⅡ型内含子剪接[15],因此推测该基因可能与早实基因的剪接有关联。
2.3 不同品种核桃matK基因片段推导的氨基酸序列及遗传距离分析
对22个不同品种来源的matK序列推导得到氨基酸序列,经BLASTP比对分析显示其属于matK超基因家族。进一步分析表明(见图2)。除薄壳香外,其他21个品种核桃均编码234个氨基酸,而薄壳香经两次克隆测序结果均在175号位置翻译终止;在这22条序列中只发生10个氨基酸位点的差异,体现在早实类群的扎343、薄壳香、金薄丰1号、西扶1号和辽核1号的序列。
运用DNAstar软件对22个核桃品种matK氨基酸序列进行多重比较,并生成遗传距离图谱(见图3)。



图2 22核桃品种matK基因氨基酸序列同源性比较Fig 2 Similarity comparation of amino acid sequences of matKs of 22 walnut species

图3 基于matK序列的22个品种核桃遗传距离图谱Fig.3 Genetic distance of 22 walnut species base on matK squences
发现22个核桃品种的种间遗传距离分化系数为0.1~0.8,百分比为99.2%~100%,其中晚实核桃品种礼品1、2号,晋龙1、2号以及清香之间的遗传距离最小,分化系数约为0.13,百分比约为99.9%;而扎343与各品种的分化系数最大,分化系数平均为0.65,百分比平均为99.3%;金薄香系列中的金薄香3号及6号与其他品种间遗传距离较大,分化系数约为0.39,百分比约为99.62%;薄壳香与其他品种的分化系数约为0.32,百分比约为99.65%;西扶1号、辽核1号与其他品种间遗传距离分化系数分别为0.5和0.53,百分比为99.53%和99.5%;而其他早实品种的遗传距离分化系数大约在0.23,百分比接近99.8%。
2.4 不同品种核桃基于matK基因片段推导的氨基酸序列种内进化关系分析
利用MEGA5.0分析软件,基于上述推导的22个核桃品种matK基因氨基酸序列分析得到NJ-系统树,如图4A所示:从中可看到5个组群,金薄香3号、清香、金薄香4号、晋龙1号、中林1号、金薄香2号、金薄香7号、晋龙2号、礼品2号、金薄香1号、金薄香6号、香玲、京861、981、金薄香8号以及礼品1号、西林3号各为一组群,得到40%的bootstap值支持;薄壳香与金薄丰1号归为一组,得到83%的bootstap值支持;辽核1号与西扶1号归为一组,得到89%的bootstap值支持;扎343自成一组。
同时还利用最大简约法构建MP-系统树,如图4B所示,22个核桃品种为一单系群,分为5个组群,其中扎343与金薄丰1号各自独立为一分支;礼品1号、礼品2号、金薄香2号、金薄香8号、晋龙1号、晋龙2号及981、西林3号为一支,但仅得到7%的bootstap值支持;金薄香3号、金薄香6号、薄壳香、香玲、辽核1号以及西扶1号聚为一支,其中金薄香6号与金薄香3号聚为一亚支,得到41%的bootstap值支持;辽核1号与西扶1号聚为一亚支,得到99%的bootstap值支持;清香、中林3号、金薄香4号、金薄香1号、京861、金薄香7号聚为一支,得到60%的bootstap值支持。
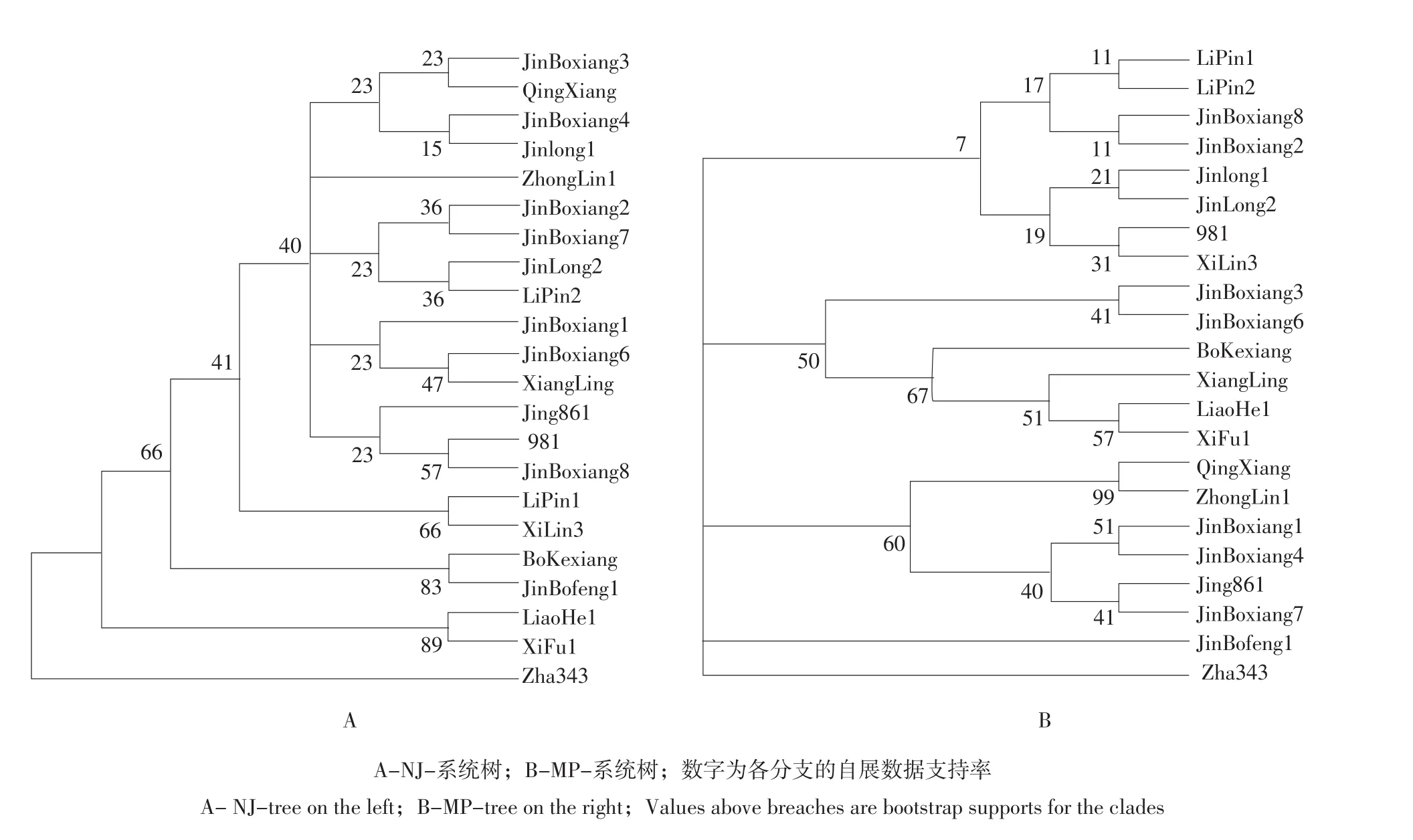
图4 基于matK序列分析得到的NJ-系统树和MP-系统树Fig.4 NJ-tree and the MP-tree base on matK sequences analysis
这两种方法建立的系统树虽然都把22个核桃品种分为5个组群,但组群划分的相同之处只有两点:一是均支持将扎343自立为一个组群,该品种是新疆林业科学院研究人员从阿克苏地区扎木台试验站早实实生核桃中选育,是我国首批早实核桃新品种,本试验结果也说明该资源的特殊性;二是均支持把辽核1号和西扶1号作为一个组群或亚支,前者是由辽宁经济林研究所经华北核桃和新疆核桃人工杂交得到的早实品种,后者是由西北林学院1981年从陕西扶风隔年核桃实生树中选育。结合供试各品种来源,如礼品1号和2号是由辽宁经济林研究所从新疆引入的纸皮核桃实生选育的两个单系,两者相似性应更多;晋龙1号和2号来源于汾阳当地的晚实核桃类群,选育单位主要是山西省林业科学研究院。只有MP-系统树将它们聚在同一亚支。有研究也表明,最大简约法适用于亲缘关系较近的类群或差异较小的序列之间的分析[15],本试验研究的是种间关系,其基因序列变异率较小,结合两系统树结果,以最大简约法构树的组群划分更合理。
3 讨论
自2009年陆地植物核心条码候选片段公布之后,研究者在条形码领域进行多项研究。于文光等运用matK序列分析法,探讨西伯利亚蓼系统学地位,并对西伯利亚蓼属及何首乌属作了界定[16];符小敏测定枇杷属4种植物及2个外类群matK基因,确立枇杷属与石斑木属亲缘关系近,将香花枇杷和大花枇杷辨别开[15];章群对杜仲matK序列分析发现其特有序列GAC,可科学鉴定杜仲真伪[13];罗集鹏等研究发现在matK基因上广藿香、土藿香及近缘藿香存在较大序列差异[17],解决广藿香与土藿香药材的鉴别问题;基于matK序列条形码技术,在天南星科多属植株、芸香科多属多种植物的鉴定,以及首乌、石斛属等植物研究中也取得一定成果。
该技术在木本类植物研究中应用较少但前景可观。现已利用matK基因系列对松科和松属植物[18]进行系统发育研究,认为银杉属与云杉属亲缘关系较近,白皮松应作为一个单系,且与亚洲地区的松属植物的亲缘关系较远;施苏华利用matK序列对进香树科及其近缘植物进行了研究[19];闫化学研究表明,matK基因可作为柑橘及其近缘植物的条形码[20]。高婷以matK和其他基因组合,运用matK-trnK序列对不同地区的菊花与野生近缘物种构建系统发育树[21];于杰等将matK和rbcL DNA序列组合,使柑橘种间水平鉴定率比单用matK更高[22]。
本文中该基因核苷酸序列有望在早、晚实核桃类群鉴定中得到应用,加速育种进程;而在不同种间关系研究只能划分遗传差异较大者,可为品种筛选提供初步信息,今后应在核基因组中寻找合适的基因与matK组合,为品种鉴定和筛选提供更精确的信息;本研究发现该基因在核桃研究中作用不仅体现在系统发育研究方面,薄壳香品种的提前翻译终止突变是否与该品种的某些特性有关,有待深入研究。
4 结论
通过克隆获得22个核桃品种matK基因近3′端长度为726 bp的EST序列,核苷酸序列分析表明,在选定的品种中早实品种发生变异,而晚实品种表现稳定,该规律是否对所有早、晚实类群适用有待于进一步研究;推导的氨基酸序列中薄壳香出现提前翻译终止,结合该基因功能推测其可能与某些早实性状功能表达有关联;而基于该基因信息建立的两类系统树均把供试的22个核桃品种划分为五个组群,且都支持扎343自立为一组群,表明扎343的特殊性。早实品种辽核1号、西扶1号在进化树上也聚为一组,说明其亲缘关系较近,但地理位置并不是造成品种差异的首要因素,这与胡桃科植物的RAPD技术研究结论存在差
异[23]。
金薄丰1号是山西省近年来从汾州核桃实生后代中选育,林木品种委员会专家田间已有鉴定,本试验中发现其与其他品种间有差异,在MP树中自立为一支,然后与扎343聚为一支,在NJ树中与薄壳香先聚为一支,再与辽核1号、西扶1号聚在一起,最后与扎343聚在一起,说明这几个品种的亲缘关系较近;金薄香系列也是山西近年来新选育的品种,研究表明只有金薄香3号与其他品种有差异,该系列品种无遗传差异,可能由于选择的基因片段不够长,不足以使遗传差异小品种区分开,今后对该基因还需深入研究,继续挖掘更适合的基因作为核桃品种鉴定依据。
[1]梅立春,郭春会,刘林强,等.中美核桃业之差距与对策[J].西北农林科技大学学报,2002,30(4):79-82.
[2]韩华柏,何方.我国核桃育种的回顾和展望[J].经济林研究, 2004,22(3):45-50.
[3]齐静.中国主栽区核桃坚果品质研究[D].保定:河北农业大学, 2009:21-42.
[4]马庆国,齐静,裴东,等.16个早实核桃良种遗传多样性的FISH-AFLP分析[J].林业科学研究,2010,23(5):631-636.
[5]Wang H,Yan Y B,Zhang J.Application of ITS sequence and SSR markers to study the relationshipbetween Juglans regia and Jug⁃lans sigillata[J].Journal of Nanjing Forestry University:Natural Science Edition,2009,33(6):35-38.
[6]杨克强,马明,孙彩玲.核桃早实基因的RAPD标记及其序列分析研究[J].中国农业科学,2007,40(9):2021-2027.
[7]叶春秀,牛建新,吕建强,等.核桃早实性相关的SCAR标记在母本和F1代上的分析[J].分子植物育种,2010,8(5):971-975.
[8]Hebert P D N,Penton E H,Burns J M,et al.Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator[J].Proceedings of the National Acad⁃emy of Sciences,2004(101):14812-14817.
[9]Hollingsworth P M,Forrest J L,Little D P,etal.A DNA barcode for land plants[J].Proceedings of the National Academy of Sci⁃ences,2009,106(31):12794-12797.
[10]Araliaceae.Phylogenetic relationships of tropical lepidoptera[J]. American Journal of Botancy,1997,84:565-580.
[11]Li Y,Gao L M,Poudel R C.High universality of MatK primers for barcoding gymnosperms[J].Journal of Systematics and Evolution, 2011,49(3):169-175.
[12]Hilu K W,Liang H.The MatK gene:Sequence variation and ap⁃plication in plant systematic[J].Amer J Bot,1997,84:830-839.
[13]章群.中药杜仲原植物的分子鉴定[J].生态科学,2004,23(2): 141-143.
[14]宋艳波,吴国良,牛洪斌.改良CTAB法在核桃叶片基因组DNA提取中的应用研究[J].山西农业大学学报:自然科学版, 2011,31(2):109-113.
[15]符小敏.广东枇杷属植物的亲缘关系[D].广州:中山大学, 2006.
[16]文光,樊守金,许崇梅,等.基于叶绿体MatK基因序列分析[J].西北植物学报,2003,23(7):1169-1172.
[17]罗集鹏,曹晖,刘玉萍.广藿香与土藿香的序列分析及其分子鉴别[J].药学学报,2007,37(9):739-742.
[18]Zhang Z Y,Li D.Molecular Phylogeny of section parrya of Pinus(Pinaceae)based on chloroplast MatK gene sequence data [J].Acta Botanica Sinica,2004,46(2):171-179.
[19]施苏华.进香树科及其近缘植物MatK序列分析和系统学意义[J].生态科学学报,2003,22(2):113-115.
[20]闫化学.柑橘及其近缘属植物DNA条形码研究[D].重庆:西南大学,2010.
[21]高婷.利用DNA条形码技术鉴定药用双子叶植物-以菊科、豆科为类[D].北京:中国协和医科大学,2010.
[22]于杰,闫华学,鲁振华,等.基于MatK和rbcLDNA序列条形码鉴定柑橘其近缘属植物[J].园艺学报,2011,38(9):1733-1740.
[23]Nicese F P,Hormaza J I.Molecular characterization and genetic relatedness amog walnut genotypes base on RAPD markers[J].Eu⁃phytica,1998,101:199-206.
Analysis of walnut interspecific phylogenetic relationships based onmatKgene sequences/
SONG Yanbo1,MEI Xia1,WANG Yong2,WANG Jinsheng1,WU Guoliang3(1.School of Life Science,Shanxi Agricultural University,Taigu Shanxi 030801,China;2. Institute of Pomology,Shanxi Academy of Agricultural Sciences,Taigu Shanxi 030801,China;3.School of Horticulture,Agricultural University of Henan,Zhengzhou 450002,China)
matKgene is known as one of the core bar code segments for the candidated land plants. In this paper,we acquired thematKgenes 726 bp EST which located in the 3'end from 22 walnut varieties and explored the value of the matK gene on establishing walnut interspecies relation by the sequence analysis.The results demonstrate that the nucleotide sequence of early bearing walnutsmatKgenes' mutation has been detected,but the later mature walnuts were opposite.Amino Acid Sequence Analysis indicated that nuthin shell had promotes premature translational-termination and generates 173 amino acids because of mutations,which was extensively different the normal products 234 amino acids.Neighbours pick Neighbor-Joining and Maxium Parsimony was used to construct the system of evolutionary tree and 22 walnut varieties were divided into five groups,we concluded that Zha343 had distinct differences with other 21 walnut varieties,Jinbofeng1 gained an advantage over the other varieties among the novel varieties and Liaohe1 and xifu1 were the closest cousin.In a word,the further research inmatKgenes provide the amolecular basis for walnut germplasm resourced protection,development and application of hybridization breeding.
walnut;matK sequence;early bearing walnut;evolutionary tree
S664.1
A
1005-9369(2014)06-0024-08
时间 2014-6-11 16:11:36 [URL]http://www.cnki.net/kcms/detail/23.1391.S.20140611.1611.010.html
宋艳波,梅霞,王勇,等.基于matK基因序列的核桃种间亲缘关系分析[J].东北农业大学学报,2014,45(6):24-31.
Song Yanbo,Mei Xia,Wang Yong,et al.Analysis of walnut interspecific phylogenetic relationships based onmatKgene sequences[J].Journal of Northeast Agricultural University,2014,45(6):24-31.(in Chinese with English abstract)
2014-02-26
河南省重大科技攻关项目(092101110600);山西农业大学创新基金项目(412500)
宋艳波(1977-),女,讲师,博士,研究方向为植物生理与分子生物学。E-mail:lzysyb@126.com
*通讯作者:吴国良,教授,博士生导师,研究方向为果树种质创新、果树栽培技术改良。E-mail:walnut-wu@126.com
