UPLC-QTof-MS 测定不同提取条件下柴胡皂苷a 和柴胡皂苷d 的转化规律
2014-01-09李宗阳潘瑞乐
郭 智,彭 冰,李宗阳,潘瑞乐*
1中国医学科学院北京协和医学院药用植物研究所,北京 100193;2 首都医科大学附属北京中医医院 北京 100010
柴胡为临床的常用中药,其原植物为伞形科柴胡属植物北柴胡Bupleurum chinense DC.或狭叶柴胡Bupleurum scorzonerifolium Wild 的干燥根,具有疏散退热、疏肝解郁、升阳举陷之功效[1]。研究表明柴胡皂苷a 和柴胡皂苷d 是柴胡药材中含量高、活性好的两个主要成分,《中国药典》(2010 版)记载柴胡药材质量标准以柴胡皂苷a 和柴胡皂苷d 作为定性定量的控制指标。文献[2-4]指出柴胡皂苷a、d 结构含有醚键,遇酸或受热不稳定,因此测定柴胡皂苷a、d 含量时建议采用低温提取并加入少量碱水。本课题组前期在测定某柴胡制剂中柴胡皂苷a 和柴胡皂苷d 的含量时,发现制剂中柴胡皂苷d 未能检出,柴胡皂苷a 的含量下降了很多。相同药材采用甲醇低温提取时柴胡皂苷a、d 具有较高的含量;但采用水煮提取时,发现水提液呈酸性,柴胡皂苷a 和柴胡皂苷d 均发生了不同程度的变化。柴胡作为临床常用中药,汤剂及中成药制剂中使用很多,且大多以水煎煮法提取。以水作溶剂加热提取柴胡药材时,柴胡皂苷a、d 如何变化、变化成何种化合物及变化程度未见有文献报道。本研究利用高灵敏度的UPLCQTof-MS 对不同提取条件柴胡药材柴胡皂苷a 和柴胡皂苷d 的变化进行定性研究,以期找到柴胡药材和制剂质量控制的合理指标。
1 材料与方法
1.1 仪器与试剂
Acquity UPLC I-class 超高效液相系统(沃特世),Xervo G2 QTof 质谱系统(沃特世),Acquity 工作站(沃特世),Masslynx4.1 工作站(沃特世);Integral 3 Milli-Q 超纯水系统(默克密理博);MS105 DU电子分析天平(梅特勒);N1100 旋转蒸发仪(东京理化);KQ-250B 超声波清洗器(昆山市超声仪器有限公司)。Optima LC/MS 乙腈(赛默飞),HPLC 甲醇(赛默飞)。提取用乙醇为分析纯(北京化工厂),提取用水为市售Wahaha 纯净水。
柴胡皂苷a(SSa)、柴胡皂苷d(SSd)标准品由中国药品生物制品鉴定所提供,批号分别为110777-200406、110778- 200506;柴胡皂苷b1(SSb1)标准品由成都曼斯特生物科技有限公司提供,批号为MUST-13 091109;柴胡皂苷b2(SSb2)标准品由天津马克生物技术有限公司提供,批号为2012-1205。柴胡样品采自甘肃陇西县,经张本刚教授鉴定为植物银州柴胡B.yinchowense 的干燥根。样品标本存于中国医学科学院药用植物研究所标本馆。
1.2 仪器条件
1.2.1 色谱条件
色谱柱:Acquity UPLC BEH C18(2.1 mm ×50 mm,1.7 μm);保护柱:VanGuand BEH C18(2.1 mm×5 mm,1.7 μm),流动相:A 为乙腈,B 为0.01%甲酸水溶液,梯度洗脱程序:0~2 min,10%~30% A;2~9 min,30%~38% A;9~11 min,38%~50% A;11~12 min,50%~100% A;12~13.5 min,100%A;13.5~13.51 min,100%~10% A;13.51~15 min,10% A;流速:0.45 mL/min;进样量:1 μL;柱温35 ℃。
1.2.2 质谱条件
负离子,MSe模式;检测范围100~1500 Da;毛细管电压:3 kV,锥孔电压:45 V,裂解电压:45~55 V;锥孔气流量:50 L/h;脱溶剂气流量:800 L/h;源温:100 ℃;脱溶剂气温度350 ℃;准确质量测定采用亮氨酸-脑啡肽(Leucine-Enkephalin,m/z=554.2615)溶液做为质量锁定溶液。
在以上条件下,四种柴胡皂苷均能达到基线分离,质谱信号响应良好。
1.3 对照品溶液制备
分别精密称定柴胡皂苷a、d、b1、b2分别为2.38、2.52、1.93、2.43 mg,使用色谱甲醇定容于2 mL 容量瓶得到对照品储备液。精确吸取储备液100 μL 于25 mL 容量瓶,色谱甲醇定容后吸取2.0 mL,使用0.22 μm 微孔滤膜过滤,弃去初滤液,取续滤液,分别得SSa、SSd、SSb1、SSb2对照品工作液。
1.4 供试品溶液制备
1.4.1 提取方法设计
选择温度(30 ℃和100 ℃)、溶剂(水和乙醇)、酸碱度(酸性、中性和碱性)作为考查因素,设计不同的提取方法,见表1。
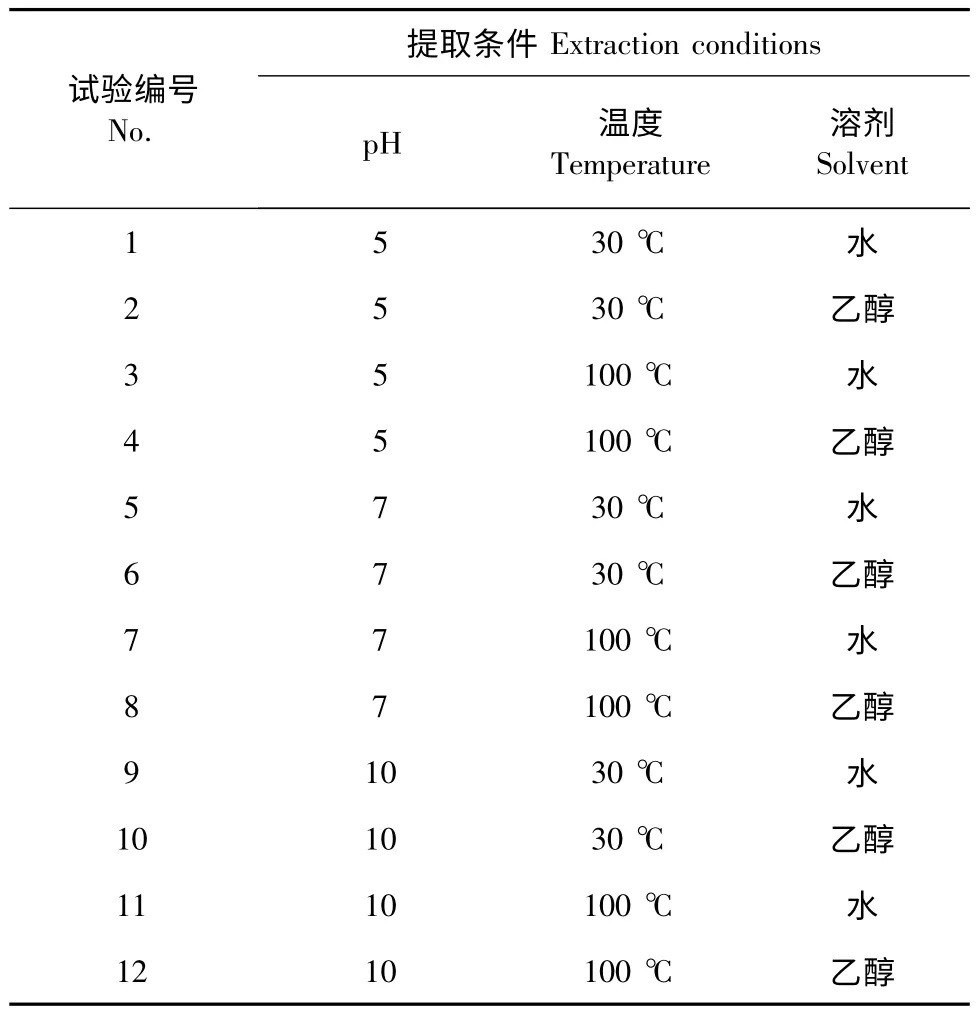
表1 柴胡提取方法设计Table 1 Different extraction conditions of saikosaponin a and saikosaponin d
1.4.2 制备流程
精密称定柴胡药材粉末(40 目)1.00 g,置于50 mL 梨形瓶中,精确加入25 mL 提取液,分别按照表2 设计的方法提取。各提取液过滤,滤渣用少量相同提取液润洗两次,合并滤液,减压回收至干,残渣加入色谱纯甲醇使其充分溶解,定溶于25 mL 容量瓶,摇匀。分析前用0.22 μm 微孔滤膜过滤,弃去初滤液,取续滤液,既得供试品溶液,编号按表1 顺序为1~12。
2 实验结果
2.1 对照品SSa、SSd、SSb1、SSb2质谱裂解规律
SSa、SSd、SSb1、SSb2属于同分异构体,在此分析条件下,SSa、SSd、SSb1、SSb2的色谱保留时间分别为6.56、10.37、8.26、6.84 min 对照品的总离子流色谱图见图1;ESI 离子源负离子模式下,它们具有基本相同的一级质谱,基峰为[M-H +HCOOH]ˉ(m/z=825.4635);二级质谱主要的碎片峰有:[M-H]ˉ(m/z=779.4594),[M-H-Glc]ˉ(m/z=617.4050),[MH-Glc-Fuc-]ˉ(m/z=471.3469),SSb1和SSb2还有[M-H-Glc-Fuc-CH2OH]ˉ(m/z=423.2360)。
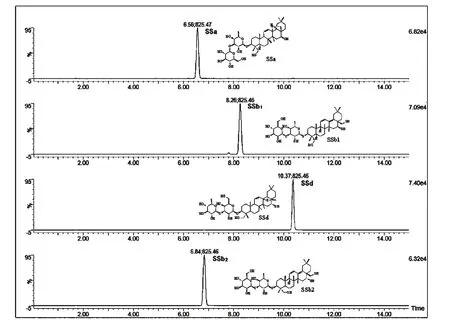
图1 对照品SSa、SSd、SSb1、SSb2的结构式和总离子流色谱图Fig.1 Chemical structures and total ion chromatograms of SSa,SSd,SSb1,SSb2standards
2.2 柴胡不同提取方法柴胡皂苷a 和d 的UPLCQTof-MS 研究
不同提取方法柴胡样品1~12 的UPLC-QTof-MS 色谱图见图2。
图2 中样品1~4 为酸性条件下提取的柴胡样品的UPLC-MS 色谱图。由图可知,四个样品均未检测出柴胡皂苷a(保留时间为6.56 min)、d(保留时间为10.37 min)。在常温酸性条件下提取的样品1和2 中检测到柴胡皂苷b1(保留时间为8.26 min)和柴胡皂苷b2(保留时间为6.84 min)。以水为溶剂,加酸加热回流条件提取的样品3 中,柴胡皂苷a、d、b1、b2均未检测出,在此条件下柴胡皂苷发生酸水解;以乙醇作溶剂,加酸加热回流提取的样品4中,可检测到脱去两分子糖的苷元加甲酸峰(m/z:517.3536)。
图2 中样品5~8 为中性条件下提取的柴胡样品的UPLC-MS 色谱图。由图可知,四个样品中均有柴胡皂苷a、d 检出。其中水、醇常温提取和醇加热提取的样品5、6、8 检测到柴胡皂苷a、d 为大量成分,基本无柴胡皂苷b1检出,有少量柴胡皂苷b2检出。由样品6 中小量柴胡皂苷b2的检出推测柴胡生药中也含有少量柴胡皂苷b2。水加热提取的样品7 中柴胡除有柴胡皂苷a、d 检出外,还有较大量的柴胡皂苷b2检出,测定其提取液的pH 值由中性转变为酸性(pH=4),由于酸性条件部分柴胡皂苷d 水解开环生成柴胡皂苷b2。
图2 中样品9~12 为碱性条件下提取的柴胡样品的UPLC-MS 色谱图。由图可知,四个样品中柴胡皂苷a、d 均为大量成分,无柴胡皂苷b1检出,四个样品均有小量柴胡皂苷b2检出。加入碱可以抑制柴胡皂苷a、d 的水解开环。
对1~12 号样品色谱图进行积分,以SSa、SSd、SSb1、SSb2的最大峰面积为基峰(100%)分别计算相对含量,图3 为1~12 号样品柴胡皂苷a、b1相对含量。

图2 样品1~12 以核质比825.46 ±0.1Da 提取离子流色谱图Fig.2 Extracted ion chromatograms (825.46 ±0.1Da)of sample1-12
图4 为1-12 号样品柴胡皂苷d、b2相对含量。
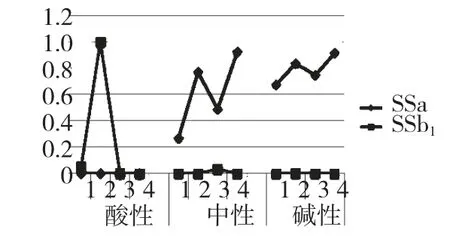
图3 样品1-12 号柴胡皂苷a、b1相对含量Fig.3 Relative contents of saikosaponin a,b1of sample 1-12
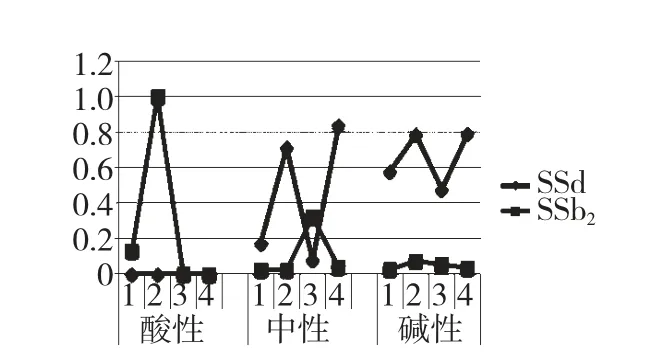
图4 样品1~12 号柴胡皂苷d、b2相对含量Fig.4 Relative contents of saikosaponin d,b2of sample 1-12
3 讨论
柴胡皂苷紫外吸收很弱或无紫外吸收,传统对柴胡皂苷的测定选择紫外检测器,使用210 nm 作为检测波长[5,6],但存在吸收较弱和干扰较多的问题。本研究选择飞行时间质谱(Q-Tof)作为检测器,解决了以上问题。同时可以给出碎片精确的分子量,有助于对化合物结构变化的跟踪和解析。前期实验通过比较正离子、负离子两种模式,发现负离子模式色谱峰型优于正离子模式,杂峰较少,故选择负离子模式。而超高效液相系统相比传统的高效液相系统能显著的缩短分析时间,提高分析效率。
本研究表明在酸性条件提取时,柴胡皂苷a 和柴胡皂苷d 全部转化。常温时柴胡皂苷a 转化为柴胡皂苷b1,柴胡皂苷d 转化为b2;加热时柴胡皂苷a 和柴胡皂苷d 发生糖苷键酸水解。在中性常温条件提取时,柴胡皂苷a、d 比较稳定,但以水作溶剂加热时,柴胡皂苷d 转化为柴胡皂苷b2。在弱碱性条件下,柴胡皂苷a 和柴胡皂苷d 均比较稳定,但四个样品仍有极小量的柴胡皂苷b2检出,但未有柴胡皂苷b1,说明柴胡皂苷b2可能是柴胡中天然存在的。
由于水作溶剂加热提取柴胡时,柴胡皂苷a、d 不稳定,并且随着加热时间的延长,柴胡皂苷a、d 会逐渐水解开环生成柴胡皂苷b1、b2。柴胡制剂多数以水加热提取,因此,柴胡制剂质量控制的指标若仍以柴胡皂苷a、d 作为指标是不科学的。柴胡制剂的质量控制建议综合考虑柴胡皂苷a、d、b1、b2总量来控制。
1 Chinese Pharmacopoeia Commission (国家药典委员会).Pharmacopoeia of the People’s Republic of China (中华人民共和国药典).Beijing:China Medical Science Press,2010.Vol I,263-264.
2 Bao Y,Li C,Shen H,et al.Determination of saikosaponin derivatives in Radix bupleuri and in pharmaceuticals of the chinese multiherb remedy xiaochaihu-tang using liquid chromatographic tandem mass spectrometry.Anal Chem,2004,76:4208-4216.
3 Liu MX(刘密新),Wu ZP(吴筑平),Yang CD(杨成对),et al.Study on saikosaponin A and D in a Chinese traditional medicine by LC/MS.J Chin Mass Spectro Soc(质谱学报),2000,21:77-78.
4 Shimizu K,Amagaya S,Ogihara Y.Structural transformation of saikosaponins by gastric juice and intestinal flora.J Pharmacobiodyn,1985,8:718-725.
5 Huang S(黄帅),Ma M(马淼),Huang QQ(黄倩倩),et al.一测多评法同步测定柴胡药材中3 种皂苷的含量.Lishizhen Med Mater Med Res(时珍国医国药),2010,4:838-840.
6 Fu Y(符颖),Zhu ZJ(朱占军),Huang S(黄帅),et al.Determination of five saikosaponins in Bupleurum yinchowense by HPLC.Nat Prod Res Dev(天然产物研究与开发),2011,3:494-497.
