新型高效液相色谱流动相手性添加剂法拆分头孢氨苄对映体
2013-12-22沈静茹余学红刘武林
沈静茹,王 伟,余学红,蔡 薇,张 祎,刘武林
(1 中南民族大学 化学与材料科学学院分析化学国家民委重点实验室,武汉 430074;2 中南民族大学 校医院,武汉 430074)
头孢氨苄是一种半合成的第一代口服头孢霉素类抗生素药物,可通过抑制细菌细胞壁合成而杀灭细菌,在临床上广泛用于敏感菌所致多种呼吸道、泌尿道等感染的治疗[1],但是有些人对头孢氨苄有不良过敏反应如休克,哮喘,皮疹等[2],这类常见抗生素的对映体分离研究应能有助于更好地推进研究其药效学和药代动力学.头孢氨苄的定量检测方法有液质(LC-MS)法[3],流体注射化学发光法[4]等.苏立强等[5]采用高效液相色谱-手性流动相添加剂(HPLC-CMPA)法分离头孢氨苄得到一对对映体的分离.但头孢氨苄结构(见图1)[6]中有3个手性碳,在β-内酰胺环上2个手性C1、C2共平面,这一刚性结构使其与C3形成2对对映体,而这4个对映异构体的分离,未见相关分离报道.近年来,HPLC-CMPA法拆分手性药物使用的手性包合试剂中环糊精类添加剂较为普遍,其中常用的手性流动相是环糊精(CD)类衍生物,如羟丙基-β-环糊精(HP-β-CD)、三甲基-β-环糊精(TM-β-CD)、羧甲基-β-环糊精(CM-β-CD)、阳离子-β-环糊精(Cationic-β-CD)、磺丁基醚-β-环糊精(SBE-β-CD)等[7].

图1 头孢氨苄结构式
本文将曾用于模拟酶催化[8]或抑制[9]领域的双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-CD(简称β-CD-C2)[10]作为HPLC流动相添加剂手性分离头孢氨苄,此衍生物制备简单,改善了β-环糊精的溶解性和立体选择性,多个羧基官能团与头孢氨苄作用位点更丰富,还可在水中溶解成弱酸与三乙胺溶液形成缓冲体系,无需另加缓冲溶液控制酸度恒定.以乙腈-添加剂缓冲溶液作为流动相,ODS-BP柱为固定相,在最佳条件下进行手性分离,可实现头孢氨苄4个对映异构体色谱峰的基线分离,为头孢氨苄的手性分离提供了新的拆分方法和体系.
1 实验部分
1.1 试剂和仪器
乙腈(色谱纯,天津市科密殴化学试剂有限公司),双-[6-氧-(3-脱氧柠檬酸单酯-4)]-β-CD)(自制),三乙胺(分析纯,上海金山亭化工试剂厂),超纯水(美国Moleculer超纯水机生产),头孢氨苄胶囊(浙江亚太药业股份有限公司),头孢氨苄标准品(中国药品生物制品检定所).
P230-II高效液相色谱仪(大连依利特分析仪器有限公司),pHS-3C 型酸度计(上海伟业仪器厂),0.22μm 微孔滤膜(上海市新亚净化器件厂).
1.2 色谱条件
色谱柱:SinoChrom ODS-BP(4.6mm×150mm)(大连依利特分析仪器有限公司);流动相︰添加剂缓冲液(V(添加剂水溶液)︰V(三乙胺)=11︰4)-乙腈;检测波长:310 nm;样品浓度:2.3 mmol/L;进样量:20μL;柱温:20 ℃;流速:0.6 mL/min.
1.3 药品前处理
头孢氨苄配制成28.75mmol/L储备液,据实验需要配成不同浓度,进样前经0.22 μm的微孔滤膜过滤.将不同浓度市售纯度为99.5%的三乙胺加入一系列不同浓度的β-CD-C2配制成添加剂缓冲液,经0.22μm的水系膜过滤脱气;色谱纯乙腈过滤脱气,备用.
2 结果与讨论
2.1 流动相乙腈与缓冲液的影响
2.1.1 乙腈-缓冲液溶解度范围测试
β-CD-C2-三乙胺缓冲液与有机调节剂乙腈存在溶解度控制的问题,在20℃,6.0 mmol/L添加剂,不同酸度时乙腈与添加剂在不同配比下的溶解度考察结果如表1所示.由表1可见,6.0 mmol/L添加剂条件下,当V(乙腈)︰V(添加剂)大于5︰5时,添加剂比例越大越不易沉淀,说明有机相越多,添加剂越易析出,越易沉淀.当V(乙腈)︰V(添加剂)小于5︰5(分别为4︰6,3︰7,2︰8,1︰9)时,无明显沉淀,不同pH值影响不大.

表1 添加剂在乙腈中的溶解度
2.1.2 乙腈-缓冲液配比变化对头孢氨苄分离的影响
以ODS-BP柱为固定相,β-CD-C2添加剂缓冲液(pH 8.4)-乙腈作为手性流动相,流速:0.6 mL/min,检测波长:310 nm,样品浓度:2.3 mol/L,进样量:20 μL,在乙腈体积分数为45%~15%变化范围内对头孢氨苄4个对映体进行手性分离,分离结果如图2所示.由图2可见,随着流动相中乙腈体积分数的减小,添加剂缓冲液体积分数的增大,分离度呈上升趋势,当乙腈体积分数降为25%时,最难分离的第1、2对映异构体之间开始分离,当乙腈体积分数达20%时分离度最大,Rs为3.25,此时1、2、3、4 四个对映异构体实现了基线分离,且此时流动相整体洗脱强度也最佳.之后随着乙腈体积分数的减小,分离状况和洗脱强度都减弱,故选择乙腈-缓冲液体积比为20︰80最适.

a~d)乙腈和添加剂缓冲液体积比分别为:35︰65;25︰75;20︰80;15︰85
2.2 流动相缓冲液pH值对头孢氨苄分离的影响
以ODS-BP柱为固定相,3 mmol/Lβ-CD-C2添加剂-三乙胺缓冲液和乙腈作为手性流动相,V(缓冲液)︰V(乙腈)=80︰20,流速:0.6 mL/min,检测波长:310 nm,样品浓度:2.3mmol/L,进样量:20 μL,通过改变添加剂与三乙胺体积来调节pH使之分别为8.4,8.6,8.8,9.0时对头孢氨苄进行手性分离,得到一系列分离结果见图3.

a)pH=8.4;b)pH=8.6;c)pH=8.8;d)pH=9.0
由图3可见,在pH 8.4~9.0时头孢氨苄均有分离迹象,其中较难拆分1和2两对映体,随着pH的增大分离度逐渐加大,到pH 8.8时,1和2对映体拆分接近基线,同时1、2、3、4 四个对映体实现基线分离,且洗脱强度最佳,综合4个对映体的峰形、洗脱强度、分离效果等因素,选择8.8为最适pH.
2.3 流动相添加剂浓度对头孢氨苄分离的影响
以ODS-BP柱为固定相,通过改变添加剂浓度分别为1,3,5,7,9 mmol/L ,用等浓度的三乙胺溶液分别调节为pH 8.8的缓冲液,以乙腈和β-CD-B2添加剂缓冲液作为手性流动相(V(乙腈)︰V(缓冲液)=20︰80),流速:0.6 mL/min,检测波长:310 nm,样品浓度:2.3m mol/L,进样量:20 μL,考察其对头孢氨苄手性分离的影响,结果见图4.由图4可见,以较难分离的1、2对映体为考察对象,当β-CD-C2存在时,头孢氨苄Rs>2.05;当浓度为5 mmol/L时,峰洗脱强度大、对称性好,分离度可达到Rs为4.93;随着β-CD-C2浓度的增大,对称性和分离度有所下降,同时当添加剂浓度大于10 mmol时易在乙腈中析出,综合考虑选择5 mmol/L为最适添加剂浓度.

c/(mmol·L-1)
2.4 对比试验
因β-CD-C2的合成是在双(-6-氧-丁烯二酸单酯)-β-环糊精(β-CD-A2)中间产物上用氯乙酸通过加成反应得到的,故对比试验选择用纯环糊精和中间体,与添加剂最佳条件下分离情况进行比对.以ODS-BP柱为固定相,在pH 8.8,乙腈与缓冲液体积比为20︰80的最佳条件下,以浓度为5.0 mmol/L的β-CD,β-CD-A2,β-CD-C2分别为HPLC的流动相添加剂和乙腈体系分离头孢氨苄,同时对比了β-CD-C2和溶剂水(图5c),结果见图5.如图5a所示,β-CD对头孢氨苄有一定分离迹象,但峰洗脱强度小,杂质峰较多且基线不稳;图5b中β-CD-A2的加入使4个对映体有了分离迹象,但洗脱强度不大,峰形对称性不好;图5c中β-CD-C2作为流动相添加剂时,在最佳条件下,不仅洗脱强度大,同时使头孢氨苄的4个对映体达到基线分离.说明对β-CD进行官能团修饰后生成直链带4个羧基的衍生物β-CD-C2与头孢氨苄中的羟基能产生氢键和π-π共轭等协同作用,使得头孢氨苄各单一对映体能与手性添加剂β-CD-C2形成复合物,在适宜的HPLC体系中实现分离.
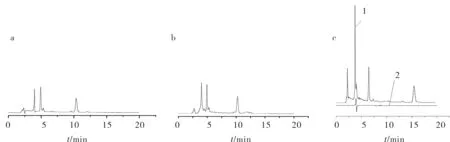
a)β-CD;b)β-CD-A2;1)β-CD-B2;2)溶剂水
3 结语
头孢氨苄是人畜最常用的抗生素之一,不可避免会在动物制品,人体血浆、肾脏等有残留,对食品安全和人体健康带来危害[11],目前对头孢氨苄的检测主要是以消旋体形式的含量检测,其结构复杂导致对映体分离困难,对映体含量检测也鲜见文献报道,本文是用新型HPLC流动相添加剂双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精作为手性流动相添加剂,建立了高效液相色谱手性拆分头孢氨苄的新方法,为单一对映体含量测定提供了可能.考察了流动相配比、pH值、手性流动相添加剂β-CD-C2浓度等因素对手性分离的影响,在pH 8.8,5 mmol/Lβ-CD-C2缓冲液中进样20 μL,V(乙腈)︰V(缓冲液)=20︰80,检测波长310 nm的最佳条件下,头孢氨苄2对对映体实现了基线分离,4个对映体色谱峰均达到基线分离,最佳条件下4峰中相邻2峰分离度Rs分别为3.41、5.34和8.93.
[1]贾亮平,张琰图,齐广才,等.高效液相色谱化学发光法检测人体血清中的头孢氨苄和甲氧苄啶[J].分析化学,2009,37(z1):99.
[2]Lina Kantiani,Marinella Farre,Josep Manuel ,et al.Development and validation of a pressurized liquid extraction liquid chromatography-electrospray-tandem mass spectrometry method forβ-lactams and sulfonamides in animal feed [J].J Chromatogr A,2010(1217):4247-4254.
[3]Cai Shengwu,Jin Lanzhang,Yan Lingqiao,et al.Simultaneous determination of 14β-lactam antibiotics in cosmetic products by liquid chromatography tandem mass spectrometry method[J].J Chromatogr B,2006(832):307-312.
[3]Sun Y,Tang Y,Yao H,et al.Potassium permanganate-glyoxal chemiluminescence system for flow injection analysis of cephalosporin antibiotics: cefalexin,cefadroxil and cefazolin sodium in pharmaceutical preparations[J].Talanta,2004,64(1):156-159.
[4]苏立强,李 冲,张晓红,等.高效液相色谱流动相添加剂法拆分头孢氨苄对映体[J].药物分析杂志,2008,28(6):921-923.
[5]国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社,2010: 208-209.
[6]尤启冬,林国强.手性药物研究与评价[M].2版.北京: 化学工业出版社,2011:200-201.
[7]杨晓为,宋发军,沈静茹,等.β-CD衍生物的反胶束微型反应器模拟酶催化氧化研究[J].分子催化,2002,16(2):111-115.
[8]徐 勇,伍 明,吴士筠,等.β-环糊精衍生物对α-羟丁酸脱氢酶的抑制作用[J].中南民族大学学报:自然科学版,2005,24(1):21-23.
[9]丁志刚,罗天顺,宋发军,等.环糊精衍生物的合成及对脲酶的抑制作用[J].催化学报,1996,17(6):567-569.
[10]倪梅林,崔晓美,殷居易,等.动物组织与水产品中头孢氨苄残留量的液相色谱-串联质谱检测[J].分析测试学报,2009,28(4):489-492.
