Improved synthesis of tenofovir disoproxil fumarate
2013-11-28MAShuaiWEIWeiJIAJingyuCAOShenghua
MA Shuai,WEI Wei,JIA Jing-yu,CAO Sheng-hua
(Sichuan Industrial Institute of Antibiotics,China National Pharmaceutical Group Corporation,Chengdu 610052,China)
Tenofovir disoproxil fumarate(1,TDF)is a potent nucleoside analogue reversetranscriptase inhibitor that is developed by Gilead Sciences,Inc.[1].It has been approved successively by US Food and Drug Administration for the treatment of HIV and chronic hepatitis B(CHB)in adult patients in 2001 and in 2008[2-3].TDF is a prodrug of tenofovir designed to improve absorption and cell permeability of the active moiety[4].Tenofovir is a nucleotide analogue of adenosine monophosphate.Its active metabolite,tenofovir diphosphate,competes with natural deoxyadenosine triphosphate for the active binding site.Incorporation of tenofovir diphosphate into viral DNA results in chain termination.Hence,reverse transcription,the key step in HIV/HBV proliferation,is inhibited[2,5].At present,TDF is widely used in clinical therapy,and in combination with emtricitabine and efavirenz to form the backbone of highly active anti-retroviral therapy[6].Many guidances recommend it as the first-line nucleoside analogue reverse transcriptase inhibitor to treat HIV and HBV.
1 SYNTHETIC ROUTE
Holy et al[7-8]has been previously reported two synthetic routes of tenofovir,both of which used D-isobutyl lactate as starting material.The two routes employed the readily available reagents,but the synthetic processes were long,and the operational processes were too cumbersome,which included adding protective groups and deprotection for several times.The yields of the two syntheses were low.Larrow J F reported a novel method to synthesize tenofovir using(±)-propylene oxide as starting material[9].However,the reactants included(salen)Cr-catalyzed and azidotrimethylsilane,which increased the cost of the route.The synthetic route was long including 8 steps,the yield was about 20%.Schutze L M and his colleague reported a new route to synthesize TDF using(S)-glycidol as starting material[10-11],the synthetic process was only 9 steps to obtain the target compound.The overall yield for this process from(R)-1,2-propylene carbonate was about 13%.Wu Wei reported a new synthetic route of TDF,which used(R)-epichlorohydrin as starting material[12].Hydrogen chloride gas and sodium hydride were used in the synthetic process,these reagents required special reaction equipments,which increased the difficult of manufacturing TDF.The total yield of this route was only 8.4%.In addition,there were many patentsand otherjournals thathave provided methods for improving the process for the manufacture of TDF at commercial scale[11,13-16].However,these methods and improvements were not obviously enhanced the yield of TDF.The synthetic route based on aforementioned process has been designed as Figure 1 and the manufacturing process has been optimized.
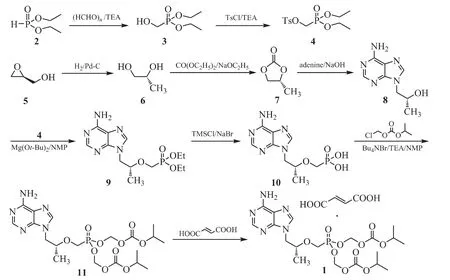
Figure 1 The designed synthetic route to tenofovir disoproxil fumarate
In this synthetic process,diethyl p-toluenesulfonyloxymethylphosphonate(4)was prepared by reacting diethyl phosphite and paraformaldehyde,and tosylating the product in situ.(S)-glycidol(5)was reduced to(R)-1,2-propanediol(6)by cataly-tic hydrogenation,which was then reacted with diethyl carbonate to afford(R)-1,2-propylene carbonate(7).The carbonate was reacted with adenine and catalyzed by sodium hydroxide to give(R)-9-(2-hydroxypropyl)adenine(8),which was then converted into tenofovir(10)including etherification with tosylate 4 and hydrolysis with trimethylsilyl chloride/sodium bromide reagent,followed by esterification with chloromethyl isopropyl carbonate to produce tenofovir disoproxil(11,TD),forming fumaric acid salt to crystallize the target compound TDF(1).
Several improvements were made to the TDF manufacturing process.Adenine reacted with the ratio of 1.2 moles compound 7,after the reaction completed,the react solution was added toluene to crystallize the product 8.The yield of this step was 68.97%.To prepare the compound 10 from 8,the processes chosed magnesium tert-butoxide as the optimum base,then utilized the lower-cost trimethylsilyl chloride/sodium bromide reagent mixture in place of trimethylsilyl bromide to hydrolyze.The two key process improvements significantly decided the total yield of the whole synthetic process.The optimum reaction molar ratio of compound 8,4,magnesium tert-butoxide and trimethylsilyl chloride/sodium bromide was 1∶1.3∶2∶4.The total yield of the two steps was improved to 62.54%,which was higherthan the documents reported about 36.3%[11],45.1%[15].The optimum reaction conditions to synthetize the compound 11 were that the molar ratio of tenofovir,tetrabutylammonium bromide,chloromethyl isopropyl carbonate and triethylamine was 1∶1∶4∶4.The introduction of tetra-butylammonium bromide obviously enhanced the reaction rate,the reaction completion was shortened to 6 h.The crude product of 11 needed not further purification,which was directly treated with fumaric acid to crystallize the target compound TDF.As a result of these changes,the total yield from adenine was improved to 30.4%,which was higher than the reported about 8.4%~21.0%[9,11-13,15].The reagents of the process were lower-cost and readily available,the reactions were moderate and easy to operate.The process improvements can be used in the manufacture of TDF at commercial scale.
2 EXPERIMENTAL SECTION
The purity of samples were analyzed by HPLC(Agilent 1200LC).1H-NMR spectra were recorded on Bruker AM-300 spectrometer in CDCl3using tetramethylsilane as the internal standard.Chemical shifts were reported as δ values(ppm).ESI mass spectrawereobtained by Agilent LC/MS/MS(1200HPLC/6410B QQQ).
2.1 Diethyl p-toluenesulfonyloxymethylphosphonate(4)
Diethyl phosphite(2,5.0 g,36.21 mmol),paraformaldehyde(1.41 g,47.07 mmol)and triethylamine(1 mL)were added in toluene(15 mL)and stirred.The reaction mixture was gradually heated to 105 ℃,and refluxed for about 3 h.The resulting mixture was cooled to 0℃,and added ptoluenesulfonyl chloride(6.21 g,32.59 mmol)and toluene(10 mL).Triethylamine(7 mL)was added slowly and the mixture was stirred for 2 h.Then the reaction mixture was warmed to ambient temperature and stirred for 12 h.The salts produced in the process were filtered and the cake was washed by toluene,the combined organic phases were washed with 5% aqueous sodium carbonate solution and brine in sequence,then dried with anhydrous sodium sulfate,filtered and concentrated under reduced pressure to afford colourless oil 7.19 g,yield was 61.6%.ESI-MS m/z:323.1[M+H]+.1H-NMR(CDCl3)δ:7.76- 7.79(d,J=8.3 Hz,2H,Ar-H),7.33- 7.36(d,J=8.1 Hz,2H,Ar-H),4.07-4.18(m,6H,2×OCH2CH3,OCH2P),2.44(s,3H,CH3),1.27-1.32(t,6H,2 × CH2CH3).
2.2(R)-1,2-Propanediol(6)
To a solution of(S)-glycidol(5,10.0 g,135 mmol)in ethanol(50 mL)was added 20% sodium hydroxide solution(2.5 g)and stirred at 0 ℃.5%Palladium on activated carbon(0.5 g)was added,and hydrogen gas was introduced to the reaction mixture.The reaction was held about 6 h until hydrogen consumption stopped.The reaction mixture was filtered with diatomaceous earth,and the filtrate was concentrated under reduced pressure to afford colourless oil,the crude product was used directly in the next step without further purification.ESI-MS m/z:77.1[M+H]+.1H-NMR(DMSO-d6)δ:4.39-4.48(m,2H,CH2CH),3.54-3.57(m,1H,OH),3.34-3.35(m,1H,OH),3.13-3.18(m,1H,CH2CH),0.98-1.00(d,J=6.2 Hz,3H,CH3).
2.3(R)-1,2-Propylene carbonate(7)
A solution of sodium ethoxide(0.22 g)in ethanol(1 mL)was added in diethyl carbonate(9.17 g,75.6 mmol),and stirred over 10 min.Then the crude product above compound 6(5.0 g,65.7 mmol)was added dropwise,the reaction mixture was heated to 100 ℃ to distill off the ethanol.The reaction was monitored by TLC,after about 6 h the reaction was completed.The reaction mass was quenched with water(50 mL),and extracted by dichloromethane,the combined organic layer was dried over anhydrous sodium sulfate and concentrated.The residue was distilled at 120 ℃ under 2 kPa to give the compound 7 as colourless oil.ESI-MS m/z: 103.2[M+H]+.1H-NMR(CD3OD)δ:4.85-4.96(m,1H,CH2CHCH3),4.04- 4.10(t,2H,OCH2CH),1.43-1.45(d,J=6.3 Hz,3H,CH3).
2.4(R)-9-(2-Hydroxypropyl)adenine(8)
Adenine(10.0 g,74.0 mmol)and sodium hydroxide(0.30 g,7.4 mmol)were added in DMF(60 mL).(R)-1,2-propylene carbonate(9.07 g,88.8 mmol)was added to the reaction mass and stirred over 10 min.The mixture was heated to 120℃ and the reaction was monitored by HPLC.When the content of adenine was below 1%,the resulting mixture was cooled to 90℃,and then toluene(60 mL)was added,much precipitation was observed.The reaction mass was continued cooled to 0℃ and held at this temperature for about 2 h.The precipitation was filtrated and the cake was washed with toluene(20 mL)chilled to 0℃.The cake was dried under vaccum to provide white solid 9.86 g,yield was 68.97%,purity was 98.2%.HPLC conditions:Diamonsil-C18column(250 mm×4.6 mm,5 μm particles),UV detection at 260 nm,mobile phase 80 mL of phosphate buffer adjusted to pH 5.5 mixed with 10 mL methanol,flow rate 1.0 mL·min-1,column temperature 40 ℃.ESI-MS m/z:194.2[M+H]+.1H-NMR(CD3OD)δ:8.20(s,1H,CH=N),8.09(s,1H,CH=N),4.25-4.29(d,J=10.7 Hz,2H,NCH2CH),4.05-4.16(m,1H,CH2CHCH3),1.20- 1.22(d,J=6.0 Hz,3H,CH3).
2.5(R)-9-[2-(Diethylphosphonomethoxy)propyl]adenine(9)
To a solution of 8(10.0 g,51.76 mmol)in N-methyl-2-pyrrolidone(50 mL)was added magnesium tert-butoxide(17.65 g,103.51 mmol)and then heated to 70 ℃.Compound 4(21.69 g,67.28 mmol)was added dropwise to the stirred mixture.The reaction was detected by TLC.After stirred about 7 h,the reaction was completed.The mixture was cooled to ambient temperature,adjusted pH to 6 with acetic acid,and then diluted with ethyl acetate(300 mL).The mixture was stirred and heated to 50 ℃ to precipitate magnesium salts.After stirred for about 30 min,the mixture was cooled,the precipitation was filtered.The cake was washed by ethyl acetate,all filtrates were combined and concentrated.Finally light yellow liquid was obtained.The crude was used in the next step directly withoutfurtherpurification.ESI-MS m/z:344.2[M+H]+.1H-NMR(DMSO-d6)δ:8.13(s,1H,N=CHN),8.05(s,1H,N=CHN),7.19(s,2H,NH2),4.24-4.27(m,4H,2 ×OCH2CH3),3.93-3.94(m,2H,NCH2CH),3.86-3.90(m,2H,OCH2P),3.28-3.32(m,1H,CH2CHO),1.08-1.21(m,9H,2 ×OCH2CH3,CHCH3).
2.6 Tenofovir(10)
The crude solution above(9)was cooled to 0℃.Sodium bromide(21.3 g,0.21 mol)was added and stirred.Trimethylsilyl chloride(22.49 g,0.21 mol)was dropwise added,then the reaction mixture was heated to 75℃.The reaction was completed at about 20 h.Then the reaction mixture was cooled to ambient temperature,quenched with water(100 mL)and extracted with ethyl acetate.The aqueous layer was cooled to about 0℃and the pH was adjusted to 3 with 40%sodium hydroxide solution.The remaining mixture was stirred for about 2 h to crystallize and then filtrated.The solids were washed with chilled water and dried to give the white solid 9.88 g,the yield was 62.54%,purity was 98.6%.HPLC conditions:Diamonsil-C18column(250 mm × 4.6 mm,5 μm particles),UV detection at 260 nm,linear gradient elution H2O with 0.05%(V/V)formic acid mixed with acetonitrile from 95∶5 to 45∶55 over 18 min,flow rate 1.0 mL·min-1,column temperature 40 ℃.ESI-MS m/z:288.1[M+H]+.1H-NMR(D2O)δ:8.39-8.40(d,J=2.1 Hz,2H,2 × CH=N),4.27-4.52(m,2H,NCH2CH),3.98- 4.01(m,2H,OCH2P),3.45-3.52(m,1H,CH2CHO),1.18-1.20(d,J=7.2 Hz,3H,CH3).
2.7 Tenofovir disoproxil(11,TD)
Compound 10(3.7 g,12.1 mmol)and triethylamine(4.9 g,48.5 mmol)was added in N-methyl-2-pyrrolidone(20 mL)and stirred.Tetrabutylammonium bromide(3.91 g,12.1 mmol)was added,and the resulting mixture was heated to 50℃.Chloromethyl isopropyl carbonate(7.4 g,48.49 mmol)was added dropwise,and then stirred for about 6 h.The reaction was monitored by HPLC for completion.The mixture was cooled to ambient temperature and added water(50 mL),and then extracted with ethyl acetate.The combined organic extracts were washed two times with saturated brine.The resulting organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure to get the light yellow solid 5.9 g.The crude product was used in the next step directly without further purification.HPLC conditions:Diamonsil-C18column(250 mm ×4.6 mm,5 μm particles),UV detection at 260 nm,linear gradient elution phosphate buffer adjusted to pH 2.5 mixed with acetonitrile from 85∶15 to 30∶70 over 15 min,flow rate 1.0 mL·min-1,column temperature 40 ℃.ESI-MS m/z:520.2[M+H]+.1H-NMR(CD3OD)δ:8.21(s,1H,N=CHN),8.12(s,1H,N=CHN),5.48- 5.62(m,4H,2 ×OCH2O),4.86-4.91[m,2H,2 ×CH(CH3)2],4.35(dd,J=3.0,13.8 Hz,1H,NCH2),4.23(dd,J=7.1,14.0 Hz,1H,NCH2),3.99-4.03(m,2H,OCH2P),3.86- 3.89(dd,J=9.2,13.2 Hz,1H,CH2CHO),1.27- 1.29[d,J=14.3 Hz,12H,2 ×CH(CH3)2],1.20-1.22(d,J=7.1 Hz,3H,CH3).
2.8 Tenofovir disoproxil fumarate(1)
To a solution of crude 11(5.9 g,11.36 mmol)in 2-propanol(20 mL)was added fumaric acid(1.58 g,13.63 mmol),the mixture was heated to 50 ℃ and stirred for about 2 h.After cooling at 0 ℃,the reac-tion mixture was stirred to crystallize.The crystals were filtrated and washed with chilled 2-propanol to 0℃.The solids were dried under vaccum to provide the white powder 5.44 g.The yield was 70.6%from 10,purity was 99.1%(HPLC conditions were the same as the sample 11).ESI-MS m/z:520.2[M+H]+(M was TD).1H-NMR(DMSO-d6)δ:13.12(br s,2H,2 × COOH),8.13(s,1H,CH),8.02(s,1H,CH),7.20(s,2H,NH2),6.63(s,2H,CH=CH),5.50-5.56(m,4H,2 × OCH2O),4.79-4.84[m,2H,2 ×CH(CH3)2],4.17- 4.24(m,2H,CH2P),3.96-3.99(m,2H,CH2N),3.31-3.33(m,1H,CH2CHO),1.22-1.24[m,12H,2 ×CH(CH3)2],1.06(d,J=6.8 Hz,3H,CH3).
Reference:
[1]ARIMILLI M N,KIM C U,DOUGHERTY J,et al.Synthesis,in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)propyl]adenine(PMPA)prodrugs[J].Antiviral Chem Chemother,1997,8(6):557-564.
[2]FUNG H B,STONE E A,PIACENTI F J.Tenofovir disoproxil fumarate:a nucleotide reverse transcriptase inhibitor for the treatment of HIV infection[J].Clin Ther,2002,24(10):1515-1548.
[3]JENH A M,PHAM P A.Tenofovir disoproxil fumarate in the treatment of chronic hepatitis B[J].Expert Rev Anti Infect Ther,2010,8(10):1079-1092.
[4]FRIIS G J,BUNDGAARD H.Prodrugs of phosphates and phosphonates:novel lipophilic α-acyloxyalkyl ester derivatives of phosphate-or phosphonate containing drugs masking the negative charges of these groups[J].Eur J Pharm Sci,1996,4(1):49-59.
[5]JONES J,COLQUITT J,SHEPHERD J,et al.Tenofovir disoproxil fumarate for the treatment of chronic hepatitis B infection[J].Health Technol Assess,2010,14(Suppl.1):23-30.
[6]YENI P.Update on HAART in HIV[J].J Hepatol,2006,44(Suppl.1):S100- S103.
[7]HOLY A,MASOJIDKOVA M.Synthesis of enantiomeric N-(2-phosphonomethoxypropyl)derivatives of purine and pyrimidine bases.Ⅰ.The stepwise approach[J].Collect Czech Chem Commun,1995,60(7):1196-1212.
[8]HOLY A,DVORAKOVA H,MASOJIDKOVA M.Synthesis of enantiomeric N-(2-phosphonomethoxypropyl)derivatives of purine and pyrimidine bases.Ⅱ.The synthon approach[J].Collect Czech Chem Commun,1995,60(8):1390-1409.
[9]LARROW J F,SCHAUS S E,JACOBSEN E N.Kinetic resolution of terminal expoxides via highly regioselective and enantioselective ring opening with TMSN3.An efficient,catalytic route to 1,2-amino alcohols[J].J Am Chem Soc,1996,118(31):7420-7421.
[10]SCHUTZE L M,CHAPMAN H H,DUBREE N J P,et al.Practical synthesis of the anti-HIV drug,PMPA[J].Tetrahedron Lett,1998,39(14):1853-1856.
[11]MUNGER J D,ROHLOFF J C,SCHUTZE L M,et al.Nucleotideanalog composition and synthesis method:US,5935946[P].1999-08-10.
[12]武卫,唐磊,邓银来,等.富马酸泰诺福韦酯新合成路线[J].中国药科大学学报,2010,41(6):505-507.
[13]RAO V U M,PRASAD V S R,BABU B R,et al.An improved process for the preparation of tenofovir:WO,2008007392[P].2008-01-17.
[14]DEBASHISH D,SHANKER R,RAO V L,et al.An improved process for the preparation of tenofovir disoproxil fumarate:WO,2011111074[P].2011-09-15.
[15]吴燕,陈国华,胡杨,等.富马酸泰诺福韦酯的合成[J].中国医药工业杂志,2011,42(2):81-83.
[16]HOUGHTON S R,MELTON J,FORTUNAK J,et al.Rapid,mild method for phosphonate diester hydrolysis:development of a one-pot synthesis of tenofovir disoproxil fumarate from tenofovir diethyl ester[J].Tetrahedron,2010,66(41):8137-8144.
