高通量DNA测序数据压缩研究进展
2013-11-26朱泽轩张永朋尤著宏
朱泽轩,张永朋,尤著宏,姜 亮,纪 震
1)深圳市嵌入式系统设计重点实验室,深圳大学计算机与软件学院,深圳518060;2)深圳大学生命科学学院,深圳518060
自1977年Sanger[1]发明基于荧光标记的末端终止法DNA测序技术以来,DNA测序技术已被广泛用于生物和医学领域.2005年Margulies等[2]提出基于循环阵列合成的高通量测序方法,又称“下一代测序(next generation sequencing,NGS)”方法.该方法已在各种实现平台上获得了超大量的DNA序列数据.采用非光学显微镜成像[3]或纳米孔技术[4]的单分子测序技术有可能改变人们研究生命蓝图的方式.测序成本的降低和测序效率的提高,促使人们不断加强对DNA测序的大规模研究,使得DNA序列数据在近年呈爆炸性增长[5](图1).现在,全球最大的DNA序列数据中心EBI和NCBI已分别存储了超过万亿个碱基对(base pair,bp).许多大型DNA研究项目,如千人基因组计划(1000 Genome Project)、国际癌症基因组计划(International Cancer Genome Project,ICGC)、DNA 元件百科全书计划(Encyclopedia of DNA Elements,ENCODE)等,正以前所未有的速度产生海量DNA序列.如千人基因组项目采用NGS测序技术,在建立后6个月内即获得了超过NCBI中心Genbank数据库累积21年的数据量.计算机是存储和处理DNA数据的主要工具,其微处理器性能和存储设备容量平均18~24个月翻一番,而DNA测序数据平均4~5个月就翻一番,DNA测序数据的增长速度已远超计算机微处理器和存储设备的增长速度.如何存储和传输高通量DNA测序产生的数据,成为决定DNA研究发展的重要因素之一.数据压缩是有效解决DNA序列存储和传输的一种重要方法.

图1 DNA测序成本和序列数据量变化趋势Fig.1 Changing trends of DNA sequencing cost and data size
1 传统DNA序列压缩方法
人类迄今对DNA序列的功能仍未完全掌握,因此在压缩过程必需保证原始数据的可靠性和信息的完整性,即在客观上要求无损压缩.DNA序列通常表示为碱基 {A,C,G,T}构成的字符串,常见压缩工具未考虑DNA序列的生物学特性,对其数据的压缩效果不甚理想.
研究DNA序列的专用压缩算法始于1993年,Grumbach等[6]提出针对DNA序列特有的互补回文结构的压缩方法BioCompress,该方法用Fibonacci编码对DNA序列中的直接重复和互补回文片段进行压缩,可有效降低这部分数据的冗余,开启了DNA序列压缩的研究之门.此后,针对DNA序列的压缩逐渐引起人们的注意,研究人员提出诸如替代法(又称字典法)和统计法[7]等.
替代法寻找重复出现频率高的DNA片段并将其编入字典,在原文中将对应片段替换为字典索引编号或使用更简短的编码方式,达到压缩目的.该方法在寻找重复片段的过程充分考虑了DNA互补回文和近似匹配等生物学特性.经典的替代压缩算法 包 括 BioCompress[6]、 CTW-LZ[8]和DNACompress[9]等.统计法主要通过统计各个碱基符号出现的概率建立预测模型,然后利用预测模型对DNA序列进行预测并采用算数编码进行压缩.代表性的统计压缩算法包括 CDNA[10]和XM[11]等.张丽霞等[12]提出的多重压缩技术属该类型.
Giancarlo等[7]对传统 DNA压缩法做了综述,指出上述DNA序列压缩方法只利用DNA序列自身的冗余信息进行压缩,对小规模DNA测序数据的压缩效果较好,但高通量DNA测序往往是对全基因组的大规模测序,使用上述方法后的压缩效率有限,需要专门针对高通量测序数据的压缩技术.
2 高通量DNA测序数据压缩
现阶段高通量DNA测序数据压缩研究主要针对全基因组测序数据.现有的高通量DNA测序方法主要基于NGS技术,常用的NGS平台包括Illumina的Genome Analyzer、罗氏454基因测序仪以及AB Life Technologies的Ion Proton.NGS测序产生的是短读(short read,长度通常在几十到几百个碱基对的DNA短片段)和质量分数(quality score,每个测定的碱基提供一个质量分数表明该测定的可信度).在测序过程中为保证数据的可靠性,需使短读对目标序列保持几十倍的覆盖率,因此,NGS测序产生的数据量远大于传统Sanger测序.质量分数因其对短读拼接(assembly)、纠错及其他后续相关研究的重要作用,也必须妥善存储.根据短读拼接和组装模式不同,NGS可分为重测序(resequencing)[13]和从头测序(de novo sequencing)[14].
2.1 重测序DNA序列压缩
重测序主要针对已完成测序物种的不同个体进行测序.由于疾病研究、设计标记、人类单核酸多态(single nucleotide polymorphism,SNP)研究等原因,需对相同物种的不同个体测序.若该物种某些个体已取得完整基因组序列,则重测序技术可将已完成的基因组作为参考样本,利用NGS技术产生大规模DNA短读高度覆盖目标基因组,然后通过短读与参考基因组的映射获得目标基因组数据.
重测序数据压缩主要通过记录短读与参考基因组的差异信息进行压缩,该方法也称为基于参考基因组压缩(reference-based compression).由于同源物种基因组之间具有高度相似性,针对重测序数据的压缩通常能达到很高的压缩比.以人类为例,任何两个人的基因组有超过99%的内容是完全相同的,若已获得参考基因组,则对于目标基因组的储存,只需存储1%额外的差异信息[15].
2.1.1 基于参考基因组压缩流程及关键技术
图2为针对NGS测序短读的基于参考基因组压缩流程,具体步骤为
1)选定一个适当的参考基因组作为压缩参考样本,通常选用具有高度相似性的同源物种序列作为参考基因组.
2)通过基于生物学特性的映射(mapping)过程,确定短读与参考基因组的匹配位置、匹配类型、差异位置、差异类型和差异内容.
3)对映射结果采用高效编码进行压缩.
重测序数据压缩涉及几项关键技术包括短读映射、映射结果编码和基因组自身压缩.
短读映射.NGS技术的一个基本特点是测序产生大量短读,重测序过程中的难题之一是短读映射,即将短读与参考基因组进行序列比对(alignment),找到每条短读在基因组中的位置和与参考基因组的差异信息.传统的映射方法如BLAT[16]或BLAST[17]主要用于Sanger测序,并不适合高通量测序数据.目前针对高通量测序数据的映射算法,主要是基于哈希表的SFE(seed filter extend)[18]算法和基于BWT(burrows wheeler transform)的映射算法[19].

图2 基于参考基因组压缩流程图Fig.2 Flow chart of reference-based compression
映射结果压缩编码.通过映射,可获得短读在参考基因组上的匹配位置、匹配类型、差异位置、差异类型及差异内容[20],这些映射结果可用有效的编码进行压缩存储.例如,已知序列由碱基 {A,C,G,T}构成,仅考虑SNP的情况,则短读映射结果可归纳如表1.

表1 映射结果Table1 Mapping results
图2列举了直接匹配、镜像匹配和互补回文3种短读类型,根据映射结果对短读进行编码,其长度会比直接编码短读每个字符明显减少.目前映射结果常用 Golomb、Delta、Elias、Gamma、MOV或Huffman等整数编码进行压缩.为提高压缩率,匹配位置和差异位置一般使用相对位置[20].使用具有生物学特征的匹配类型如镜像重复、配对重复和互补回文也有助于提高数据压缩比[21].
压缩参考基因组.重测序数据压缩和解压都必需使用参考基因组.有些物种,如人类,其参考基因组本身就有几Gbit,为便于存储和传输,必需考虑对参考基因组自身进行压缩.
2005-2011年,高通量DNA数据压缩有了长足进步[22-26].其中,Korodi等[22-23]提出基于统计法的NML算法,首次尝试对大型基因组整体压缩,并提出 GeNML算法.其后,Kuruppu等[24-25]提出基于字典法的RLZ算法,并进行改进.Christley等[15]使用各种不同编码技术和外部SNP信息将完整人类基因组压缩至4 Mbit大小.
2.1.2 代表算法
随着NGS技术的不断发展,对于重测序数据压缩的研究广受关注,表2汇集了近年提出的代表算法及其主要特点.表中所有算法都是基于参考基因组的压缩方法.其中,Quip、Cramtools和NGC主要针对短读,而DNAzip、BWB和GRS是针对完整目标基因组的压缩.这些方法各有千秋,在实际使用中,应该充分考虑压缩对象、参考基因组和其他辅助数据的可获得性,以及计算资源来选择恰当的压缩方法.
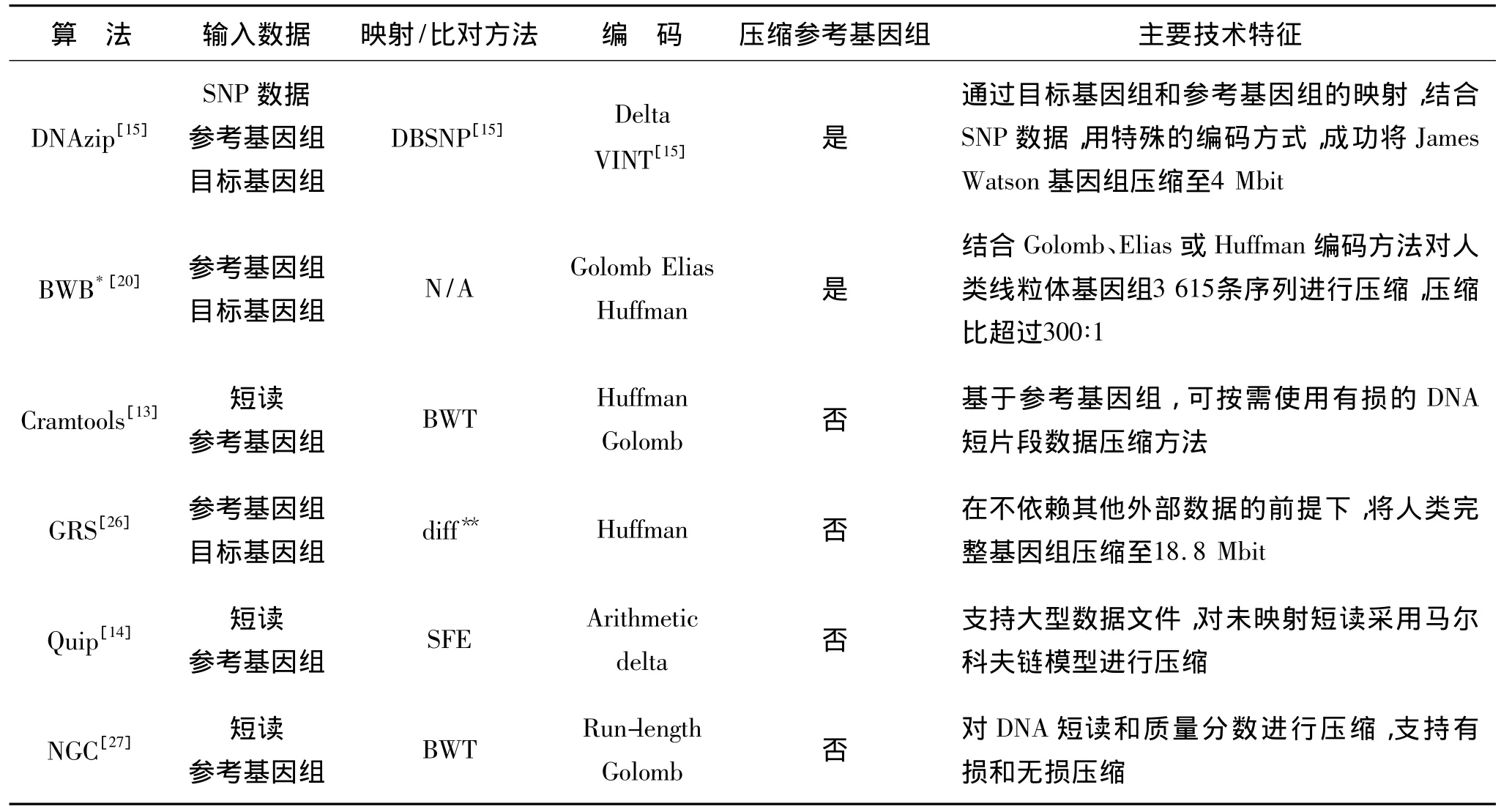
表2 重测序数据压缩代表算法Table2 The state-of-the-art compression algorithms of resequencing data
基于参考基因组的压缩方法压缩比很高,但其使用亦有明显局限,对于参考序列依赖性太强,但有些测序数据并不存在现成的参考基因组;另外,由于压缩和解压缩都需要相同的参考基因组,参考基因组必须事先保存在本地,如果参考基因组缺失将直接影响压缩数据的使用.
2.2 从头测序数据压缩
与重测序不同,从头测序不依赖现成的参考基因序列,而是直接对待测个体进行测序,然后对测序产生的短读进行从头拼接和组装.因此,针对从头测序数据的压缩也无外部参考序列的支持.对从头测序数据的压缩研究仍处于起步阶段,目前其压缩流程主要分两个阶段:①用一小部分短读拼接成叠连群(contigs)作为临时参考基因组;②采用基于参考基因组压缩方法依次进行短读映射,映射结果压缩编码.
由于压缩过程中使用的参考基因组不是来自外部而是由部分短读拼接获得,可见其关键步骤是短读拼接,即通过短读再现完整DNA序列的过程.NGS产生的大部分短读存在重叠区域,这为合并短读提供了必要信息.目前,NGS短读拼接算法主要有 OLC(overlap layout consensus) 算 法[27]、de Bruijn图算法[28]和基于OLC或 de Bruijn图的贪婪算法[29].除短读拼接,从头测序数据压缩的其他技术与重测序数据压缩类似.
目前针对从头测序数据压缩的研究相对较少,代表性的有Fritz等[13]提出的对无法映射短读采用从头测序数据压缩方法和Jones等[14]提出基于拼接的从头测序压缩算法Quip.该算法使用概率数据结构极大减少了使用de Bruijn图进行拼接时的内存需求,有效提高了短读拼接的规模,为以后从头测序数据压缩算法的发展提供参照.
与重测序压缩相比,从头测序压缩优势在于它不依赖于外部参考基因组,自完备性好,但它在一定程度上仍受制于拼接技术.从头测序压缩方法也适用于重测序数据,但其压缩比低于重测序压缩方法,如果已获得适当的参考基因组,则应优先考虑重测序压缩方法.
2.3 质量分数的压缩
质量分数伴随着短读序列的产生,每一个碱基对对应一个质量分数,质量分数的保存有利于序列数据的后续分析和使用,因此质量分数的压缩存储也非常重要.与DNA序列只由4种碱基构成不同,质量分数由更多字符表示,另外质量分数通常拥有很多的冗余信息,对其编码通常会有很高的熵,因此质量分数压缩较DNA序列压缩更难[14].目前质量分数压缩有无损和有损两种方法.
对质量分数的无损压缩主要采用Huffman编码、LZ77和马尔科夫链技术.Tembe等[30]使用Huffman编码对质量分数进行压缩,Deorowicz等[31]将数据中不同类型的质量分数采用不同的编码方式,如哈希表、游程编码和Huffman编码进行压缩.Jones等[14]使用三阶马尔科夫链对质量分数进行压缩.
文献[13,31-32]提出了对质量分数的有损压缩方法,如用随机化凑整的方法减少描述质量分数的字符,匹配到参考基因组后丢弃质量分数,对质量分数量化等.有损压缩可以提高压缩比,但也丢失了部分信息,因此采用这种压缩方式值得商榷.
2.4 压缩数据检索
压缩数据可减少存储空间和传输时间,但对压缩数据进行分析前需进行解压.随着测序数据量的增大,解压变得十分耗费计算资源,解压后的数据需被加载入内存进行分析,这对内存容量也提出了更高要求.如果能让DNA序列在压缩状态下直接查询、提取和检索,将有效减少资源消耗.目前基于压缩序列数据的索引技术主要有:Burrows-Wheeler索引、压缩后缀数组和LZ索引[33].
Burrows-Wheeler索引.FM-index[34]被认为是第一个构建Burrows-Wheeler索引的算法,使用新的数据结构用于搜索和索引,可促进压缩序列上搜索功能的实施.Makinen等[35]提出FM-index的改进版,使RLFM在空间和时间复杂度方面有较大提升.
压缩后缀数组.Grossi等[36]提出的CSA方法,使用压缩后缀数组构建压缩后缀树和空间有效的索引机制.RLCSA算法[37]是CSA的改进版,其在压缩和索引性能上都有一定提升,并允许索引多序列集合.
LZ索引.Ukkonen等[38]尝试用LZ77算法查询压缩数据,使用LZ77压缩字符串引发k-mer索引.Kreft等[39]提出的LZ-End索引使用LZ77算法解析输入字符串,在索引高度重复的序列数据中有一定优势.
在压缩状态实现序列的常规分析是未来发展的主要趋势.上述索引算法可在不解压的情况下有效提取和搜索序列子串,为以后压缩基因组的分析与应用提供帮助.
展 望
目前,高通量DNA测序数据的压缩研究已取得一定成果,但其在计算资源、压缩算法和测序平台方面仍面临巨大挑战[40].DNA测序输出的数据量和计算机资源之间的缺口正在不断加大,处理时间过长已成为DNA测序数据分析不可避免的问题.结合云计算平台的压缩和分析技术[41],有望缓解计算压力.
未来DNA测序平台会产生更优质的短读,如第3代测序采用基于纳米孔的单分子测序方法[4],短读长度可达到1~3 kbp,压缩算法需要不断顺应DNA测序技术的发展,提供压缩序列的检索功能,适应不同应用需求.压缩算法需进一步提高效率,可采用平行化策略减少计算机内存消耗,如GSQZ[30]算法.
不同测序平台产生不同质量品质和长度的高通量短读,对压缩算法的选择也有很大的影响,因此需要统一标准,评价不同平台的数据质量.
DNA测序技术不断推动生物工程和医学等相关领域的发展[42],在高速和低价的前提下,为医学研究和诊断学等领域的广泛应用提供了可能.基因组学、功能基因组学和新病毒机理等领域的进步均得益于DNA测序的发展[43-45].解决好高通量DNA测序数据存储和传输问题是推动这些领域进一步发展的前提.未来DNA序列的数据量可能超乎想象,为迎接挑战,需发展全新的DNA序列压缩算法.
/References:
[1]Sanger F,Nicklen S,Coulson A R.DNA sequencing with chain-terminating inhibitors[J].Proceedings of the National Academy of Sciences of the United States of A-merica,1977,74(12):5463-5467.
[2]Margulies M,Egholm M,Altman W E,et al.Genome sequencing in microfabricated high-density picolitre reactors [J].Nature,2005,437(7057):376-380.
[3]Kai A.STM and AFM of bio/organic molecules and structures[J].Surface Science Reports,1996,26(8):263-332.
[4]Hibbs X,Krstic S,Mastrangelo A,et al.The potential and challenges of nanopore sequencing[J].Nature Biotechnology,2008,26(10):1146-1153.
[5]Kahn S D.On the future of genomic data[J].Science,2011,331(6018):728-729.
[6]Grumbach S,Tahi F.Compression of DNA sequences[C]//In Proceedings of Data Compression Conference.Snowbird(USA):IEEE Computer Society,1993:340-350.
[7]Giancarlo R,Scaturro D,Utro F.Textual data compression in computational biology:asynopsis[J].Bioinformatics,2009,25(13):1575-1586.
[8]Matsumoto T,Sadakane K,Imai H.Biological sequence compression algorithms[C]//Proceedings of Genome Informatics Workshop,Tokyo,2000:43-52.
[9]Chen X,Li M,Ma B,et al.DNA compress:fast and effective DNA sequence compression [J].Bioinformatics,2002,18(2):1696-1698.
[10]Loewenstern D,Yianilos P N.Significantly lower entropy estimates for natural DNA sequences[J].Computational Biology,1999,6(1):125-142.
[11]Cao M D,Dix T I,Allison L,et al.A simple statistical algorithm for biological sequence compression[C]//In Proceedings of the Conference on Data Compression,Snowbird(USA):IEEE Computer Society,2007:43-52.
[12]Zhang Lixia, Song Hongzhi. Multiple-compression of DNA sequence data[J].Journal of Computer Applications,2010,30(5):1379-1382.(in Chinese)张丽霞,宋鸿陟.多重压缩DNA序列数据 [J].计算机应用,2007,30(5):1379-1382.
[13]Fritz M H Y,Leinonen R,Cochrane G,et al.Efficient storage of high throughput DNA sequencing data using reference-based compression [J].Genome Research,2011,21(5):734-740.
[14]Jones D,Ruzzo W,Peng X,et al.Compression of nextgeneration Sequencing reads aided by highly efficient de novo assembly[J].Nucleic Acids Research,2012,40(22):e171.
[15]Christley S,Lu Y,Li C,et al.Human genomes as email attachments[J].Bioinformatics,2009,25(2):274-275.
[16]Kent W J.BLAT:the BLAST-like alignment tool[J].Genome Research,2002,12(4):656-664.
[17]Altschul S,Gish W,Miller W,et al.Basic local alignment search tool[J].Journal of Molecular Biology,1990,215(3):403-410.
[18]Li H,Ruan J,Durbin R.Mapping short DNA sequencing reads and calling variants using mapping quality scores[J].Genome Research,2008,18(11):1851-1858.
[19]Li H,Durbin R.Fast and accurate short read alignment with burrows-wheelertransform [J].Bioinformatics,2009,25(14):1754-1760.
[20]Brandon M C,Wallace D C,Baldi P.Data structures and compression algorithms for genomic sequence data[J].Bioinformatics,2009,25(14):1731-1738.
[21]Zhu Z,Zhou J,Ji Z,et al.DNA sequence compression using adaptive particle swarm optimization-based memetic algorithm [J].IEEE Transactions on Evolutionary Computation,2011,15(5):643-658.
[22]Korodi G,Tabus I.An efficient normalized maximum likelihood algorithm for DNA sequence compression [J].ACM Transactions on Information Systems,2005,23(1):3-34.
[23]Korodi G,Tabus I.Normalized maximum likelihood model of order-1 for the compression of DNA sequences[C]// Data Compression Conference. Snowbird(USA):IEEE Computer Society,2007:33-42.
[24]Kuruppu S,Puglisi S J,Zobel J.Relative Lempel-Ziv compression ofgenomes forlarge-scale storage and retrieval[C]//in proceedings of the 17th International Symposium on String Processing and Information Retrieval.Los Cabos:Springer,2010:201-206.
[25]Kuruppu S,Puglisi S J,Zobel J.Optimized relative Lempel-Ziv compression of genomes[C]//Proceedings of Australasian Computer Science Conference.Perth(Australia):Australasian Computer Science,2011:91-98.
[26]Wang C,Zhang D.A novel compression tool for efficient storage of genome resequencing data[J].Nucleic Acids Research,2011,39(7):E45-U74.
[27]Miller J R,Koren S,Sutton G.Assembly algorithms for next generation sequencing data [J].Genomics,2010,95(6):315-327.
[28]Pevzner P A,Tang H,Waterman M S.An Eulerian path approach to DNA fragment assembly [J].PNAS,2001,98(17):9748-9753.
[29]Li R Q,Zhu H M,Ruan J,et al.De novo assembly of human genomes with massively parallel short read sequencing [J].Genome Research,2010,20(2),265-272.
[30]Tembe W,Lowey J,Suh E.G-SQZ:compact encoding of genomic sequence and quality data[J].Bioinformatics,2010,26(17):2192-2194.
[31]Deorowicz S,Grabowski S.Compression of DNA sequence reads in FASTQ format[J].Bioinformatics,20 11,27(6):860-862.
[32]Popitsch N,Haeseler A V.NGC:lossless and lossy compression of aligned high throughput sequencing data[J].Nucleic Acids Research,2012,41(1):e27.
[33]Kuruppu S S.Compression of Large DNA Databases[D].Melbourne:Melbourne School of Engineering,2012.
[34]FerraginaP,Manzini G.Opportunistic data structures with applications[C]//Proceedings of the 41st Annual Symposium on Foundations of Computer Science.Redondo Beach:IEEE Computer Society,2000:390-398.
[35]Makinen V,Navarro G.Succinct suffix arrays based on run-length encoding[C]//In Proceedings of the 16th Annual Symposium on Combinatorial Pattern Matching.Jeju Island(Korea):Springer,2005:40-66.
[36]Grossi R,Vitter J S.Compressed suffix arrays and suffix trees with applications to text indexing and string matching[J].SIAM Journal on Computing,2005,35(2):378-407.
[37]Siren J,Valimaki N,Makinen V,et al.Run-length compressed indexes are superior for highly repetitive sequence collections[C]//String Processing and Information Retrieval.Berlin:Springer,2009:164-175.
[38]Karkkainen E,Ukkonen.Lempel-Ziv parsing and sublinearsize index structures for string matching[C]//Proceedings of the 3rd South American Workshop on String Processing.Belo Horizonte(Brazil):World Scientific Publishing Company Incorporated,1996:141-155.
[39]Kreft S,Navarro G.Self-Indexing based on LZ77 [C]//In Proceedings of the 22th Annual Symposium on Combinational PatternMatching. Palermo(Italy):Springer,2011:41-54.
[40]Bao S,Jiang R,Kwan W,et al.Evaluation of nextgeneration sequencing software in mapping and assembly[J].Journal of Human Genetics,2011,56(9):406-414.
[41]SteinL D.The case for cloud computing in genome informatics [J].Genome Biology,2010,11(5):207.
[42]Ansorge W J.Next-generation DNA sequencing techniques[J].New Biotechnology,2009,25(4):195-203.
[43]Horner D S,Pavesi G,Castrignano T,et al.Bioinformatics approaches for genomics and post genomics applications of next generation sequencing[J].Briefings in Bioinformatics,2010,11(2):181-197.
[44]Morozova O,Marra M A.Applications of next-generation sequencing technologies in functional genomics[J].Genomics,92(5):255-264.
[45]Yang X,Charlebois P,Gnerre S,et al.De novo assembly ofhighly diverse viralpopulations[J].BMC Genomics,2012,13:475.
