在镁合金阳极氧化膜上的化学电泳涂装
2013-10-27胡波年何晓梅张德春宋政伟
胡波年,何晓梅,张德春,宋政伟,余 刚
(1.湖南工学院 材料与化工系,湖南 衡阳 421002;2.湖南大学 化学化工学院,湖南 长沙 410082)
镁合金是一种新型的材料,具有密度小、强度高、电磁屏蔽好等特点,在通信、汽车、电子等行业具有很大的应用潜力[1-2].但是,镁合金的耐蚀性极差,这大大限制了它大范围的应用,如何提其耐蚀性成为进一步推广应用所要解决的首要问题[3].目前,镁合金的防腐措施主要有金属镀层[4-7],化 学 转 化 膜[8-10]、阳 极 氧 化[11-12]、离 子 植入[13-14]等.但是,这几种方法均各有一些缺陷.化学转化膜制备方法简单,但是膜的致密性差,容易开裂,无法阻止镁合金基底的腐蚀;阳极氧化能够在镁合金表面形成多孔的氧化膜,阻止镁合金基底的进一步腐蚀,但是其工艺复杂,耗能较大;电镀和化学镀的镀层一般分为阳极镀层和阴极镀层,最常见的镁合金防腐的金属镀层为镍磷合金镀层,作为阴极镀层最大的缺点就是如果镀层存在微孔将加速镁合金基底的腐蚀,基底在很短时间内产生穿孔,且施镀步骤复杂,耗能大[15].
有机涂层在镁合金的防腐方面具有很大的潜力,相比其他防腐措施,有机涂层具有花费低、性能好等特点[16],近年来,较新的镁合金有机涂层有富镁涂层[17-19],超疏水涂层[20],电泳涂层.电泳涂层作为有机涂层的一种,与其他有机涂层制备方法相比具有耗能少、成膜时间短、设备简单及对样品形状要求不高等特点,在汽车行业电泳漆作为底漆被广泛应用[21].但是,在电泳过程中对电泳的电压控制较为麻烦,也增加了电泳的成本.宋光铃[22-25]等通过实验发现,在不加外加电场的情况下,镁合金在环氧型阴极电泳漆中能够自发形成一层很薄的化学电泳漆层,能够大大提高镁合金的耐蚀性.研究发现镁合金表面化学电泳涂层耐蚀性与镁合金表面状态有很大关系,将镁合金试样用砂纸打磨,质量分数为10%的HCl酸洗1 min后吹干,然后350℃处理40min.再与电泳漆反应10s后,镁合金表面涂层厚度可达1.8~2.0 μm,反应24h后涂层厚度为13μm.涂层形成的关键是当镁合金放入电泳漆中时,镁合金表面能产生足够多的氢氧根离子,氢氧根与阳离子树脂反应生成不溶于水的树脂沉积在试样表面形成涂层.按照宋光铃所述工艺,酸洗后镁合金表面会形成较薄的氢氧化镁层,与氧化镁相比,相同质量的氢氧化镁碱性较小,涂层的厚度受氢氧根的扩散速率控制,当氢氧根浓度较低时,涂层的厚度随时间增加较慢,当350℃热处理后部分氢氧化镁转化为氧化镁,表面碱性提高,从而使涂层厚度增加.一般的电泳涂装工艺几分钟即可以获得20~40μm厚的涂层,而化学电泳涂装所得涂层厚度较薄(只有1~2μm),耐蚀性相对较低,所以化学电泳工艺需要进一步改进,以提高涂层厚度及耐蚀性.
本文拟通过简易的阳极氧化增加镁合金表面活性氧化镁的含量,提高镁合金表面碱性,通过在氯化钠溶液中低电势阳极氧化的方法,使镁合金表面形成活性氧化镁膜,为化学电泳涂层的形成提供充足的碱性环境,进而提高涂层的厚度,使其能够达到一般电泳涂层的厚度.而化学电泳涂装不用电能,显然具有节能生产的优势,为镁合金的腐蚀防护提供一种新的有效防护方法.
1 实验材料与方法
1.1 涂层的制备
实验选取AZ31B镁合金材板作涂装试样,主要成分质量分数(%)为:2.5~3.5Al,0.7~1.3 Zn,0.2~1.0Mn,0.05Si,0.01Cu,剩余的为Mg.电泳漆固含量为质量分数7%的聚氨酯型阴极电泳漆(东莞崇威化工机械有限公司).
将厚度为2mm的镁合金AZ31B平板切成2 cm×3cm的长方形作为化学电泳的试样.试样在自来水冲洗的情况下用600#的砂纸进行抛光,然后用蒸馏水冲洗,并用热风吹干.将镁合金试样为阳极,以相同大小的Cu为阴极,电极之间间隔2 cm,放入0.2mol·cm-3的氯化钠水溶液中施加200mA·cm-2的电流密度对镁合金进行表面氧化处理.处理好的镁合金试样在超声波的情况下用蒸馏水清洗,然后热风吹干,放入马弗炉350℃下处理30min,试样在常温下自然冷却.
烘烤后的镁合金试样放入电泳漆中,在室温条件下放置45min后缓慢拉出,使表面电泳漆流回漆槽.然后在室温下放置12h,使其自然风干,当试样表面的漆层完全干后,将其放入130℃的烘箱中烘烤25min后取出进行性能测定.
1.2 性能测定方法
镁合金试样为研究电极(保留1cm2的工作面,非工作面用环氧树脂密封),饱和甘汞电极作为参比电极,铂电极作为对电极,使用美国Gamry公司的Interface 1000电化学工作站进行电化学测量.
开路电势测量:开路电势可以反映镁合金表面电极电势的变化和涂料反应的变化关系.电极试样经过阳极氧化处理后,放入电泳漆中,测量电极的开路电势随时间的变化曲线.
极化曲线的测量:将含电泳漆涂层的试样浸入质量分数为3.5%的NaCl水溶液中,待电势相对稳定后,进行极化测量.极化电势范围为±200 mVOCP,扫描速度1mV·s-1.
交流阻抗的测量:分别测定阳极氧化后的镁合金试样在电泳漆中阻抗的变化和形成的电泳涂层在质量分数3.5%NaCl水溶液中交流阻抗的变化.控制交流电信号的电势振幅为5mV,测量频率范围为0.05~100 000Hz.阻抗图采用软件ZsimpWin拟合.
1.3 基底和涂层的表征
镁合金阳极氧化后表面形貌:阳极氧化经350℃热处理后氧化膜的表面形貌,以及化学电泳涂层表面及截面形貌用场发射扫描电镜(S4800,日本日立公司制造)表征.
1.4 浸泡析氢检测实验
将工作面积为1cm2试样放入质量分数为3.5%的NaCl水溶液中浸泡,以裸体镁合金作为对比,按照文献[26]的方法测定析氢速率,评定涂层的耐蚀性能.试验装置图见图1,每个实验重复三次,取平均值.
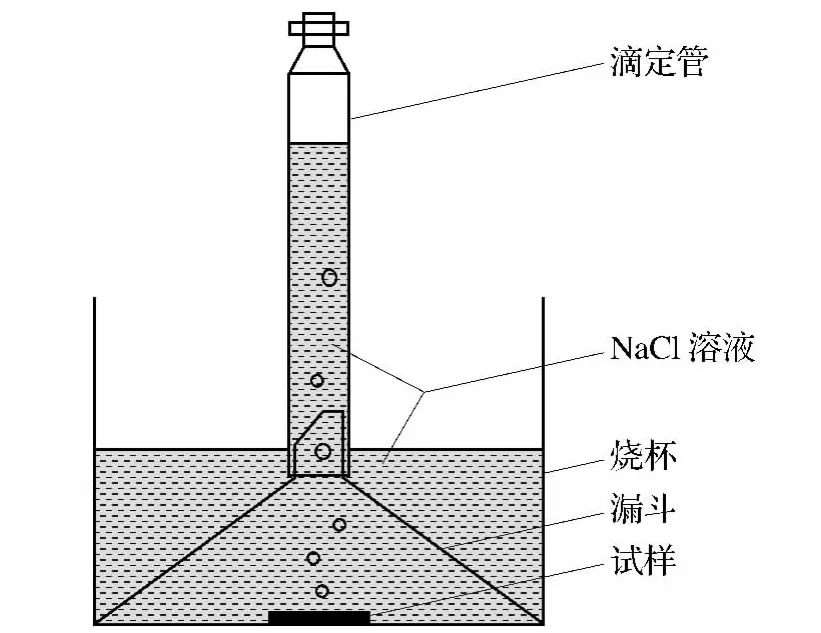
图1 析氢速率测定装置图Fig.1 Schematic diagram of a set-up for measuring hydrogen evolution
2 结果与讨论
2.1 活性氧化镁层的制备
图2a是未经过阳极氧化的AZ31B镁合金基底的形貌,镁合金基底较为平整,5万倍的内插图可以清晰地分辨出基底的α相和β相.将AZ31B镁合金试样放入0.2mol·cm-3的氯化钠电解液中用200mA·cm-2的电流密度处理1、3、5min,经过1~3min阳极氧化的镁合金基底表面还存在未完全氧化的区域,5min后则完全被氧化膜覆盖,如图2b所示.阳极氧化后的镁合金表面的氧化膜粗糙,产生0.5~1.0μm宽度的裂纹;图2c为350℃热处理30min后的氧化膜SEM形貌,与图2b对比可以看出,热处理后表面裂纹减少,氧化层表面变得更加粗糙.从图2c的内插图也可以清晰地看出氧化膜具有蜂窝状微结构,突起的片状结构尺寸约40~100nm.

图2 镁合金AZ31B基底在阳极氧化前后的表面形貌图(内插图为高倍数SEM图)Fig.2 Surface morphologies of AZ31Bsubstrate before and after anodic oxidation
从图2分析可知,350℃热处理后,脱水形成蜂窝状微结构的比表面积更大,而镁合金基底在打磨后表面与水的接触角小于90°,为亲水性表面,根据 Wenzel方程[27]:
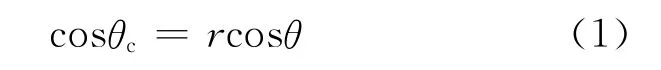
式中,r为固液接触面实际面积与几何面积的比率,θc和θ为处理后表面与处理前表面的固液接触角,处理后接触角接近0,θc<θ,可以得出r值大于1,当水滴滴到试样表面时,实际接触面积变大,亲水性增加.从实验现象也可以看出,当水滴滴到阳极氧化后的试样表面时,水能够完全浸湿,即水滴能够与镁合金表面微结构的凸凹面直接接触,表面处于Wenzel状态.
图3为阳极氧化后和经350℃热处理40min前后的表面EDX能谱,测得表面元素的质量百分比如表1所示.从表1中可以看出,阳极氧化后的氧化膜经过热处理后的表面主要成分是Mg和O两种元素,除H元素不能被检查出来,还含有少量的Cl、Al和Zn元素.热处理后表面O的百分含量有所增加,Cl的百分含量减少.热处理作用是使氢氧化镁分解为氧化镁和水,由于水的蒸发,表面出现蜂窝型微结构,增大了比表面积从而增加了表面的吸附能力,热处理同时也能使表面裸露的金属进一步氧化,从而使氧含量增加.另外,热处理也能使MgCl2发生分解,生成MgO,使氧含量增加,氯含量减少.
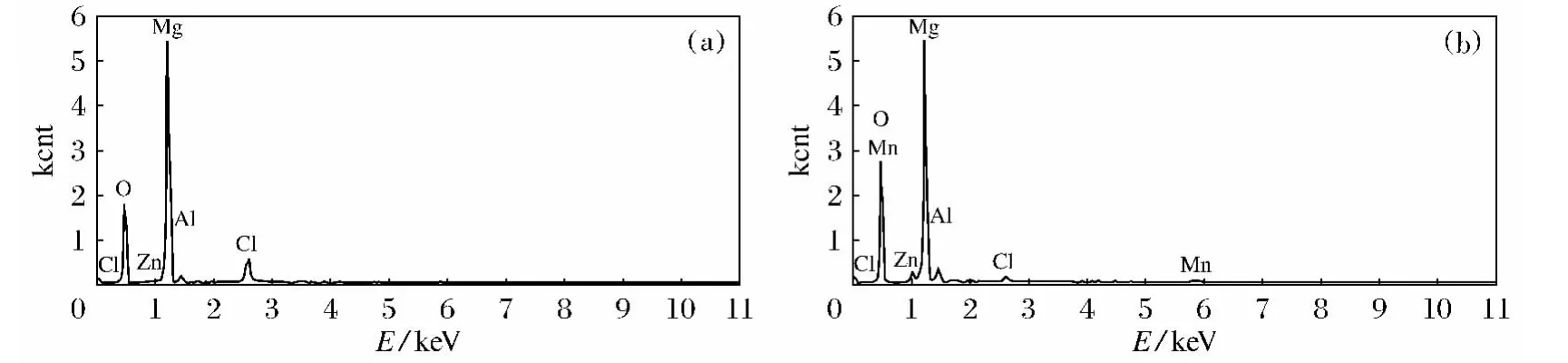
图3 阳极氧化膜热处理前后的EDX谱图Fig.3 EDX spectra of the anodic oxidation film before and after heat treatment
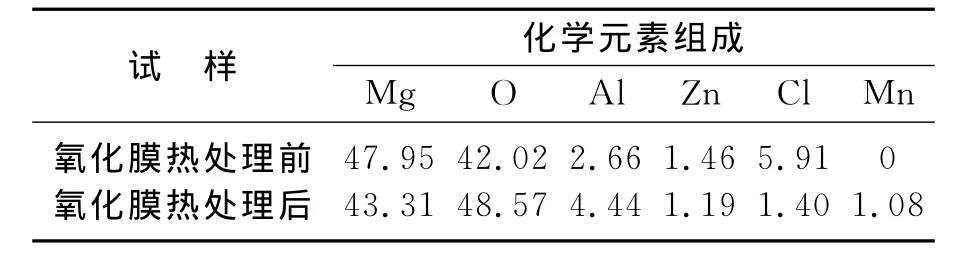
表1 阳极氧化膜热处理前后的元素组成(质量分数)Table 1 Elemental composition of anodic oxidation film before and after heat treatment(mass fraction)%
2.2 电泳涂层的形成机理
将经过5min阳极氧化后的试样放入电泳漆中,然后再把试样缓慢从电泳漆中拉出时,可以用肉眼很清晰地看到试样表面形成了一层均一的电泳漆层,几分钟后漆层变干.
通过氧化膜在电泳漆中的开路电势变化来分析化学电泳涂装的机理,图4是阳极氧化5min后的镁合金AZ31B试样在电泳漆中的开路电势随时间变化曲线.开路电势先略降后迅速上升,500s后基本平稳.由此可以看出试样表面的变化情况,当阳极氧化后的镁合金试样浸入电泳漆中时,试样表面首先出现少量的氧化膜溶解现象,开路电势下降;而后电泳漆迅速沉积在镁合金表面,开路电势上升;随着涂层的增厚,镁合金和电泳漆之间被涂层隔离,开路电势趋于稳定.
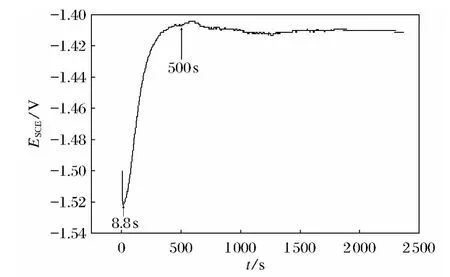
图4 镁合金试样在阳极氧化5min后放入电泳漆中的开路电势随时间变化曲线Fig.4 The dependence of OCP of the sample with anodic oxidation film after dehydration in the E-coating bath on time
AZ31B分别在电泳漆中反应5、10、15、20、25、30、35、40、45min的阻抗如图5所示.
图5a中出现一个明显的容抗弧,容抗弧半圆不完整,低频处半圆变形,容抗弧的最高点对应的频率为特征频率,特征频率能够反映出涂层的耐蚀性,特征频率越小,涂层耐蚀性越好.图5b中lgω-Φ的关系图显示出两个时间常数的特征,高频区的第一个时间常数τ1(=QcRpo)来自于涂层电容Qc以及涂层微孔电阻Rpo的贡献;低频区的第二个时间常数τ2(=QdlRdl)来自于双电层电容Qdl以及极化电阻Rdl的贡献[28-30].图5b中lgω-lg|Z|的低频区应出现平台区域,但出现一条斜线,说明电极与电泳漆之间存在扩散层,低频与高频区平台被一条直线连接,该直线与低频区平台切线的交点所对应的频率为特征频率,随着试样与电泳漆反应时间的延长,直线斜率有变大,特征频率变小,涂层的耐蚀性增加.所以阻抗的等效物理模型可以用图5c中的电路表示,其中Rs为电泳漆溶液的电阻,Rpo为涂层微孔电阻,Rdl为极化电阻,Qc为涂层电容,Qdl为双电层电容,Zw为扩散阻抗.电极的阻抗可以表示为:
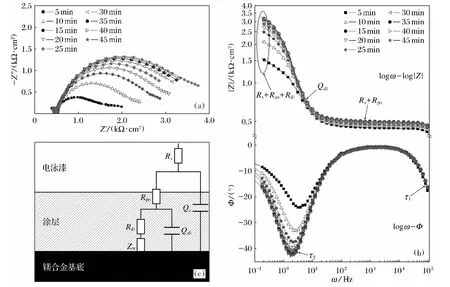
图5 AZ31B在不同反应时间下形成的涂层阻抗Fig.5 Coating impedance of AZ31Bformed at different reaction times
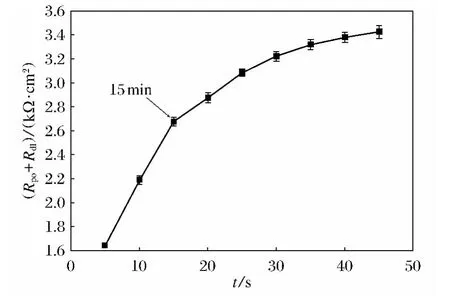
图6 涂层形成过程中电阻Rpo+Rdl随时间变化曲线Fig.6 The dependence of Rpo+Rdlsum on applying time
通过图5a、图5b的阻抗谱图结果,应用ZsimpWin软件拟合可以得出Rs、Rpo、Rdl的阻值.Rpo+Rdl阻值随时间的变化曲线如图6所示,容抗弧的直径为Rpo+Rdl,容抗弧直径随着涂装时间的延长而增大.随着反应的延长,镁合金AZ31B与电泳漆之间的电阻逐步增加,15min以前电阻增加比较快,电泳漆与镁合金表面在很短的时间内就形成一层不溶于水的聚氨酯膜.AZ31B镁合金试样在聚氨酯阴极电泳漆中放置7 min并室温干燥后的表面微观形貌如图7a所示,未热处理时涂层表面不致密,表面存在微孔结构,电泳漆层在室温下干燥过程中,涂层中的水由内向外蒸发,使涂层出现很多直径100~500nm的微孔.进一步反应过程中,氢氧根能够从镁合金表面通过涂层的微孔缓慢地向外逸出与电泳漆反应,由于氢氧根的扩散速率较慢,为反应控制步骤,所以化学电泳的成膜过程为扩散控制.随着反应的进行,膜的覆盖率越来越高,膜厚增加,氢氧根的逸出速率变慢,膜层的增长速率也随之下降,这与开路反应的情况一致.当在电泳漆中反应时间超过15min后,镁合金表面电泳漆会发生脱落现象,随着时间的增长,镁合金基底裸露出,与水反应产生氢气,由于涂层表面孔洞小,气体无法逸出,使涂层发生小范围的鼓泡甚至脱落现象.所以反应时间应控制在15min以内.

图7 AZ31B镁合金化学电泳涂层的SEM图Fig.7 SEM images of electroless E-coating formed on AZ31Bin the E-coating bath for 7min
电泳涂层经过130℃热处理后涂层表面的微观形貌如图7b所示,涂层表面无明显的孔隙,由于有机聚氨酯加热后融化,可排除一些微孔,使得AZ31B表面被一层致密的有机电泳涂层覆盖.
热处理后的涂层呈现较好的疏水性,由于镁合金腐蚀多在有水的电解质溶液中进行,而疏水涂层能够有效地隔离镁合金与腐蚀电解液的接触,起到较好的防腐效果.涂层表现出疏水性的原因一方面由于有机涂层的表面能低,水不能完全润湿涂层表面,另一方面由于表面粗糙的结构,涂层微结构中的凹陷部分被空气填充,当水滴滴到涂层表面时,水滴与涂层凹陷的部分无法接触,只能在涂层突起的部分停留.根据Cassie-Baxter方程[31]:
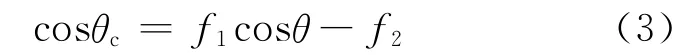
式中,θc和θ分别是聚氨酯修饰粗糙面和平滑面的固液接触角;f1和f2分别为液体与固体和空气接触面积所占的比例(f1+f2=1).镁合金基底经过阳极氧化处理后表面粗糙度增加,化学电泳后的涂层表面有细小的突起结构,当突起结构足够高时能够阻止液滴接触微结构的凹面,此时液体与涂层的接触面积小于液体与空气的接触面积,无法润湿表面.
图8是涂层的截面SEM图,图形显示有机涂层厚度为15~40μm,氧化膜厚度为7μm.涂层厚度的不均一,是由于基底的α相和β相的腐蚀不同时发生,α相先腐蚀,腐蚀较深,β相后腐蚀,腐蚀较少,基底表面形成粗糙不平的结构,涂层经过热处理后流平,凹陷部位涂层厚,突出部位涂层薄.这种凹凸不平基底有利于涂层与基底之间的机械咬合,提高涂层的结合力.

图8 复合涂层截面照片Fig.8 SEM image of the composite coating cross section
根据化学电泳的反应机理来看,反应的关键是镁合金与电泳漆的接触面具有足够的OH-以推动树脂成膜.
镁合金表面经过阳极氧化处理后形成一层活性氧化镁膜,在有水的情况下,活性氧化镁溶于水形成氢氧化镁,氢氧化镁在酸性条件下电解出氢氧根离子,促进反应的进行,主要反应方程为
许沁的抛光部在凌源,一个五层楼民房的底层。抛光部不大,就一个大车间,有几排水泥大理石砌成的台面,安放着十来台抛光机。十来个男员工戴着单薄的头罩和灰不溜秋的口罩,举着小五金,对着抛光机上的砂轮打磨。
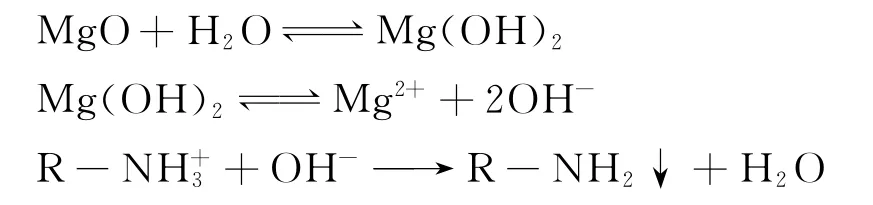
从反应方程可以看出,当镁合金表面完全被氧化膜覆盖时,与电泳漆反应过程中无气体放出,所形成的涂层理论上不会存在微孔结构.
镁合金表面阳极氧化处理后粗糙度和亲水性增加,当试样放入电泳漆中时,电泳漆能够更好地浸润试样表面,参与反应的试样表面积增大,从而增大了试样与电泳漆反应的活性.
阳极氧化的主要产物为 Mg(OH)2,但是并非所有的 Mg(OH)2都能沉积在电极镁基底表面,一部分氧化产物粘附在镁基底表面,另外一部分氧化产物会溶解在溶液中.热处理后一部分Mg(OH)2转化为MgO,作为反应主要物质的氧化镁,能够为反应提供大量的氢氧根,促进树脂的沉积,形成较厚的有机电泳涂层.所以保证在镁基底表面粘附着足够的活性MgO膜,是提高化学电泳涂装活性的关键所在.
如果镁合金表面未进行阳极氧化处理,镁基底会直接与电泳漆中水反应生成 Mg(OH)2,而阴极电泳漆pH在6.0左右,溶液为酸性,能够促进Mg(OH)2的水解,镁合金表面氢氧根浓度增加,推动化学电泳的进行.其反应机理为:

由于在反应过程中有氢气的产生,镁合金表面所形成的涂层孔洞较大,涂层不致密,对镁基底的保护性能下降.显然,镁合金基底表面形成更多的活性氧化镁膜有利于化学电泳涂装反应的进行,形成无气孔的致密电泳涂层.
2.3 化学电泳涂层耐蚀性
(1)试样耐蚀性交流阻抗谱图的表征.涂层的耐蚀性可以由其在腐蚀介质中的阻抗大小直接反映,将试样做成电极放入质量分数为3.5%的NaCl水溶液中,开路电势稳定10min后,分别测定1、3、5、7、10min化学电泳涂装形成涂层的阻抗如图9所示.
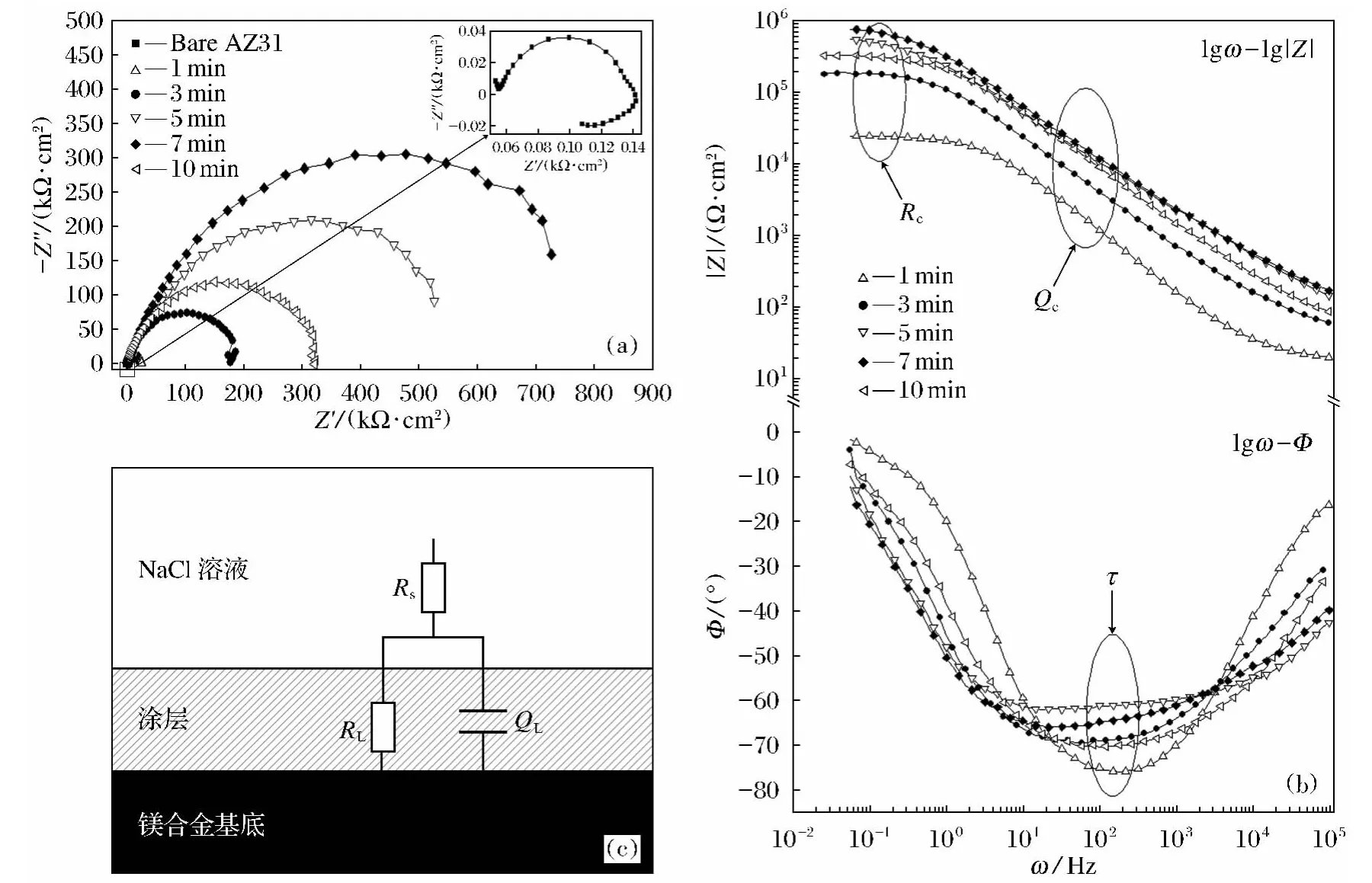
图9 不同涂装时间下获得的涂层在质量分数为3.5%的NaCl水溶液中阻抗变化Fig.9 EIS spectra measured in mass fraction of 3.5%NaCl solution and equivalent circuit for composite coating
由图9b可以看出1~10min化学电泳涂装所得涂层的阻抗呈现一个时间常数的特征,时间常数τ1来源于化学电泳层电阻RL和QL的贡献.从图9b看出,低频区出现一个平台,没有出现扩散的特征,说明镁合金表面的电泳层隔绝了镁合金基底与NaCl的接触,涂层相当于一个纯电容,电阻值很大,电容值很小.此时阻抗所对应的物理模型如图9c所示,Rs为电泳漆溶液电阻,RL为化学电泳层电阻,QL为化学电泳层电容.电极的阻抗可以表示为式(4)、式(5):
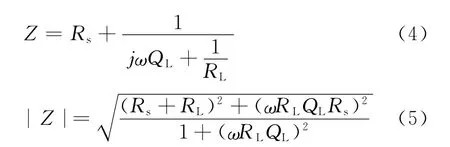
由式(5)可以看出,当频率很低时,|Z|≈Rs+RL,与频率无关,在波特图(lgω-lg|Z|)低频区出现平台.由图9a可以看出,涂装时间在1~7 min内,随着时间的增长,容抗弧直径RL增大,涂层阻抗增大,耐蚀性变好,但是涂装10min后,RL出现减小的情况,涂层阻抗变小.从图9b可以推出不同涂层阻抗的特征频率对应电阻,反应不同时间所对应的特征频率不相同,在1~7min涂装时间内随着时间的延长特征频率减小,阻抗增大,涂层耐蚀性增强,10min涂装的涂层特征频率增大,阻抗下降,涂层耐蚀性下降.
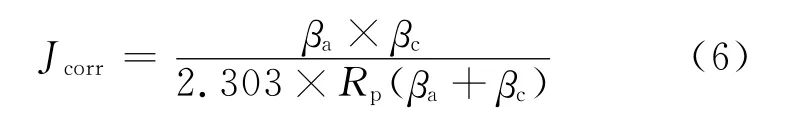
求得Jcorr(见表2),具有阳极氧化层和化学电泳层双层保护膜的镁合金比镁合金基底的腐蚀电流密度降低了至少三个数量级以上,说明该涂层能够较好地延缓镁合金基底的腐蚀速率,提高基底的耐蚀性.
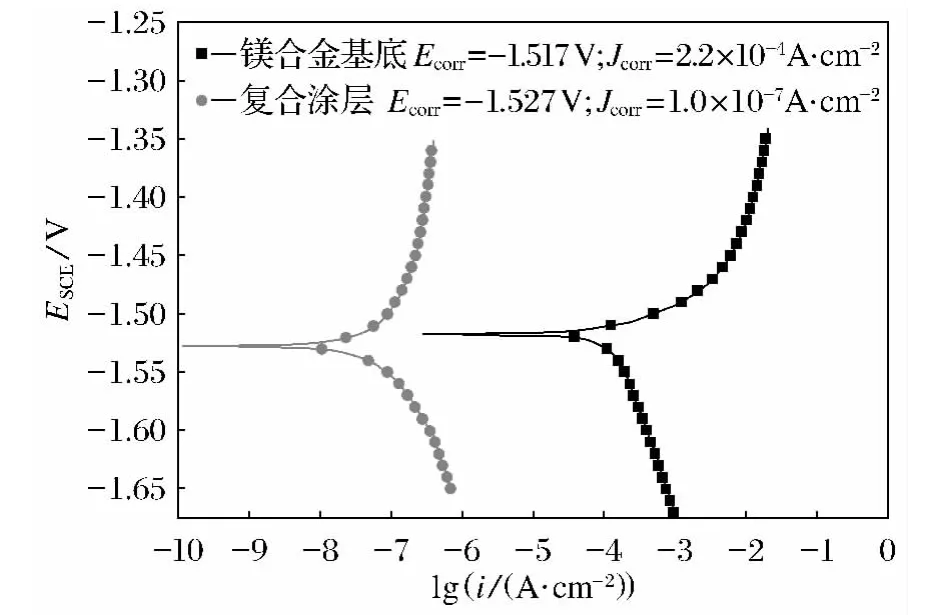
图10 裸体镁合金及化学电泳后涂层镁合金的极化曲线Fig.10 Potentiodynamic polarization curves of the bare and composite coating covered AZ31B magnesium alloy in 3.5%NaCl solution
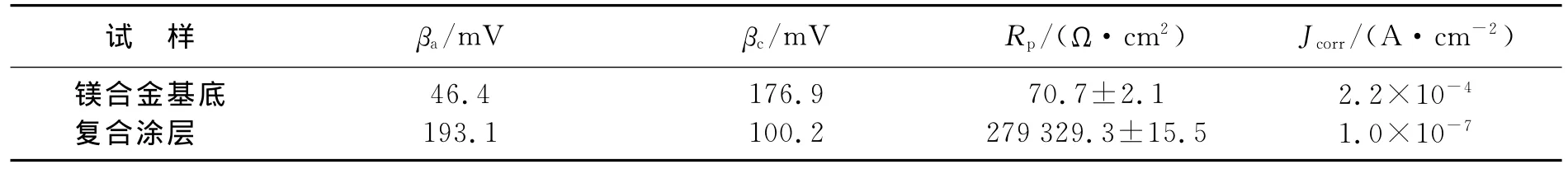
表2 裸体镁合金和化学电泳涂装后的镁合金在NaCl液中腐蚀的电化学参数Table 2 Electrochemical corrosion parameters of bare Mg alloy and composite coating coated Mg alloy in NaCl solution
2.4 不同表面前处理方法获得电泳涂层的浸泡实验结果分析
对镁合金直接化学电泳、酸洗后化学电泳和阳极氧化后化学电泳获得的涂层在质量分数为3.5%的NaCl中浸泡,测定不同时间下的腐蚀析氢体积变化.
以裸体镁合金腐蚀作为对照,每种样品做三次重复浸泡实验,实验的偏差示于图11中.裸体镁合金在NaCl水溶液中的析氢量与浸泡时间呈直线关系,说明裸体镁合金的析氢量是匀速增加的,斜率最大,表明析氢速率大;直接化学电泳后的镁合金析氢量随着时间而增加,但是析氢量均小于裸体镁合金,这是由于直接化学电泳涂装时镁基底与电泳漆在反应过程中有氢气析出,形成的涂层不致密,涂层孔洞较大,当腐蚀性介质渗入孔洞时容易发生腐蚀.
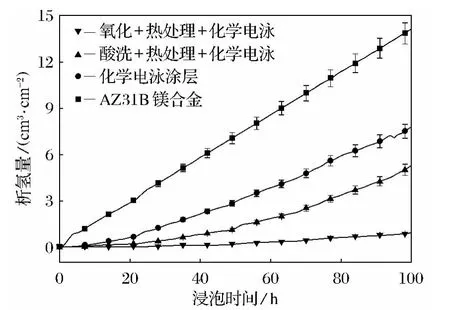
图11 不同试样在质量分数3.5%NaCl溶液中析氢量随时间变化曲线(温度18~22℃)Fig.11 The dependence of evolved hydrogen volume on immersion time in 3.5%NaCl solution in various specimens(with the temperature of 18~22℃)
镁合金酸洗后再化学电泳,形成化学电泳涂层的试样的腐蚀析氢量也是随着时间的延长而增加,但腐蚀析氢速率小于直接化学电泳镁合金试样.应用笔者开发的阳极氧化+化学电泳的工艺获得的镁合金涂层试样浸泡在质量分数为3.5%的NaCl溶液中100h后的单位试样面积析氢量不到1cm3/cm2,析氢量很小,说明镁合金的腐蚀反应很慢,比前两种前处理获得的化学电泳涂层对镁合金的保护作用好得多.
3 结 论
(1)用氯化钠溶液进行简单阳极氧化并在350℃热处理后,镁合金的阳极氧化膜微观表面具有蜂窝状结构,表面粗糙度增大,亲水性增强,氧化镁含量增多,反应活性增大.
(2)阳极氧化后化学电泳涂装的新工艺所得涂层在7min涂装反应内,厚度可达15~40μm,再延长涂装时间涂层会脱落,7min形成的化学电泳涂层表面均一,无明显的孔洞,具有较好的疏水性能.
(3)阳极氧化后化学电泳涂装所获得的复合涂层使镁合金的自腐蚀电流密度减少三个数量级以上,镁合金的耐蚀性大幅提高,比直接化学电泳涂装和酸洗后的化学电泳涂装获得的涂层的阻抗大,耐蚀性好,复合涂层对镁合金基底的保护作用好.
[1] Gray J E,Luan B.Protective Coatings on Magnesium and Its Alloys—a Critical Review[J].Journal of Alloys and Compounds,2002,336(1/2):88-113.
[2] Ferrando W A.Review of Corrosion and Corrosion Control of Magnesium Alloys and Composites[J].Journal Of Materials In Civil Engineering,1989,11(4):299-313.
[3] Song G L.Recent Progress in Corrosion and Protection of Magnesium Alloys[J].Advanced Engineering Materials,2005,7(7):563-586.
[4] Seifzadeh D,Rajabalizadeh Z.Environmentally-friendly Method for Electroless Ni-P Plating on Magnesium Alloy[J].Surface and Coatings Technology,2013,218(3):119-126.
[5] Wang Z C,Jia F,Yu L,et al.Direct Electroless Nickelboron Plating on AZ91DMagnesium Alloy[J].Surface and Coatings Technology,2012,206(17):3676-3685.
[6] Xie Z H,Yu G,Li T J,et al.Dynamic Behavior of Electroless Nickel Plating Reaction on Magnesium Alloys[J].Journal Of Coatings Technology And Research,2011,9(1):107-114.
[7] Sudagar J,Lian J S,Chen X M,et al.High Corrosion Resistance of Electroless Ni-P with Chromium-free Conversion Pre-treatments on AZ91DMagnesium Alloy[J].Transactions of Nonferrous Metals Society of China,2011,21(4):921-928.
[8] Lee Y L,Chu Y R,Chen F J,et al.Mechanism of the Formation of Stannate and Cerium Conversion Coatings on AZ91DMagnesium Alloys[J].Applied Surface Science,2013,276(7):578-585.
[9] Huang P P,Latham J A,MacFarlane Douglas R,et al.A Review of Ionic Liquid Surface Film Formation on Mg and its Alloys for Improved Corrosion Performance[J].Electrochimica Acta,2013,in press.
[10] Chen Y G,Luan B L,Song G L,et al.An Investigation of New Barium Phosphate Chemical Conversion Coating on AZ31Magnesium Alloy [J].Surface and Coatings Technology,2012,210(10):156-165.
[11] Choi Y I,Salman S,Kuroda K,et al.Synergistic Corrosion Protection for AZ31Mg Alloy by Anodizing and Stannate Post-sealing Treatments[J].Electrochimica Acta,2013,97(5):313-319.
[12] Gu Y H,Bandopadhyay S,Chen C F,et al.Effect of Oxidation Time on the Corrosion Behavior of Micro-arc Oxidation Produced AZ31Magnesium Alloys in Simulated Body Fluid[J].Journal of Alloys And Compounds,2012,543(12):109-117.
[13] Liu C L,Xin Y H,Tian X B,et al.Corrosion Behavior of AZ91Magnesium Alloy Treated by Plasma Immersion Ion Implantation and Deposition in Artificial Physiological Fluids[J].Thin Solid Films,2007,516(2/3/4):422-427.
[14] Tian X B,Wei C B,Yang S Q,et al.Corrosion Resistance Improvement of Magnesium Alloy Using Nitrogen Plasma Ion Implantation[J].Surface and Coatings Technology,2005,198(1/2/3):454-458.
[15] Song G L.“Electroless”Deposition of a Pre-film of Electrophoresis Coating and its Corrosion Resistance on a Mg Alloy[J].Electrochimica Acta,2010,55(7):2258-2268.
[16] Hu R G,Zhang S,Bu J F,et al.Recent Progress in Corrosion Protection of Magnesium Alloys by Organic Coatings[J].Progress In Organic Coatings,2012,73(2/3):129-141.
[17] Lu X Y,Zuo Y,ZhaoX Y,et al.The Improved Performance of a Mg-rich Epoxy Coating on AZ91D Magnesium Alloy by Silane Pretreatment[J].Corrosion Science,2012,60(7):165-172.
[18] Lu X Y,Zuo Y,Zhao X H,et al.The Study of a Mg-rich Epoxy Primer for Protection of AZ91DMagnesium Alloy[J].Corrosion Science,2011,53(1):153-160.
[19] Lu Xiangyu,Zuo Yu,Zhao Xuhui,et al.The Influence of Aluminum Tri-polyphosphate on the Protective Behavior of Mg-rich Epoxy Coating on AZ91DMagnesium Alloy[J].Electrochimica Acta,2013,93(3):53-64.
[20] Xu W,Song J,Sun J,et al.Rapid Fabrication of Largearea,Corrosion-resistant Superhydrophobic Mg Alloy Surfaces[J].ACS Applied Materials and Interfaces,2011,3(11):4404-4414.
[21] Luan B L,Yang D F,Liu X Y,et al.Corrosion Protection of Magnesium Alloys Using Conversion and Electrophoretic Coatings[M]∥Song G.Corrosion of Magnesium Alloys.Canada,2011:2-34.
[22] Song G L,Liu M H.The Effect of Surface Pretreatment on the Corrosion Performance of Electroless E-coating Coated AZ31[J].Corrosion Science,2012,62(9):61-72.
[23] Song G L,Liu M H.The Effect of Mg Alloy Substrate on“Electroless” E-coating Performance[J]. Corrosion Science,2011,53(11):3500-3508.
[24] Song G L.A Dipping E-coating for Mg Alloys[J].Progress In Organic Coatings,2011,70(4):252-258.
[25] Song Guangling.“Electroless”E-Coating:An Innovative Surface Treatment for Magnesium Alloys [J].Electrochemical And Solid State Letters,2009,12(10):D77-D79.
[26] Huo H W,Li Y,Wang F H.Corrosion of AZ91D Magnesium Alloy with a Chemical Conversion Coating and Electroless Nickel Layer[J].Corrosion Science,2004,46(6):1467-1477.
[27] Wenzel R N.Resistance of Solid Surfaces to Wetting by Water[J].Industrial And Engineering Chemistry,1936,28(8):988-994.
[28] Bonora P L,Defloriana F,Fedrizzi L.ElectrochemicalImpedance Spectroscopy as a Tool for Investigating Underpaint Corrosion[J].Electrochimica Acta,1996,41(7/8):1073-1082.
[29] Fedrizzi L,Rodriguez F J,Rossi S,et al.The Use of Electrochemical Techniques to Study the Corrosion Behaviour of Organic Coatings on Steel Pretreated with Sol-gel Zirconia Films[J].Electrochimica Acta,2001,46(24/25):3715-3724.
[30] Ma F.Use of Electrochemical Impedance Spectroscopy for Thestudy of Corrosion Protection by Polymer Coatings[J].Journal of Applied Electrochemistry,1995,25(3):187-202.
[31] Cassie A B D,Baxter S.Wettability of Porous Surfaces[J].Transactions of the Faraday Society,1944,40(6):546-551.
[32] Stern M,Geary A L.Electrochemical Polarization I.A Theoretical Analysis of the Shape of Polarization Curves[J].Journal of the Electrochemical Society,1957,104(1):56-63.
