功能化四氧化三铁的合成和表征及其对钙离子的吸附
2013-10-22潘志权黄齐茂
周 红,朱 明,潘志权,黄齐茂
(武汉工程大学化工与制药学院,湖北 武汉 430074)
0 引 言
磁性纳米颗粒因其优异的性能在生物、医药、催化剂等方面具有广泛用途[1-2],但由于磁性纳米颗粒在制备、分离、后处理中存在颗粒团聚、颗粒尺寸分布不均,且易被氧化等问题[3-4],必须对磁性颗粒表面进行改性[5].常用的改性方法有:直接应用引发剂进行自由基聚合反应,使其包覆于聚合物内部[6-8],或采用Stöber包覆二氧化硅再利用硅胶偶联剂如γ-氨丙基三乙氧基硅烷(APTES)对其表面进行修辞[9],该类型颗粒因引入了功能团—NH2使其易于进行表面修辞[10-12],得到的纳米材料已用于Cu(Ⅱ)吸附、抗菌、催化性能的研究[13].在上述SiO2改性的纳米颗粒中引入磁性Fe3O4核,可借助外加磁性分离使之更易分离、纯化及循环利用.另外,以丙烯酸甲酯(MA)及乙二胺(EDA)进行迈克尔加成和酰胺化反应,可合成出不同代数的树形分子[14],该树形分子中因含伯氨基、叔氨基及酰胺基,而具有螯合金属离子的功能[15].尽管人们对树形分子接枝的SiO2的颗粒及APTES改性的磁性纳米颗粒的合成、表征已作了相关研究,但以该类型树形分子修辞的磁性纳米颗粒尚未见报道[16].
水中钙离子的去除对其综合利用起着重要作用.常用的除钙方法可分为沉淀法、吸附法、溶剂萃取法、阳离子交换树脂法等.其中吸附法因具有操作简单、选择性高、净化度高等而受到关注.本研究结合磁性颗粒易分离及含功能基的树形分子的螯合性能,合成了树形分子修辞SiO2包覆的磁性纳米颗粒(合成路线见图1),并考察了该核壳磁性纳米颗粒对钙离子的吸附作用,讨论了钙与该功能基包覆的磁性纳米颗粒之间的配位关系.

图1 酯化及酰胺化核壳磁性纳米Fe3O4的合成路线Fig.1 Synthesis of esterification and amidation modified core shell magnetic nanoparticles Fe3O4
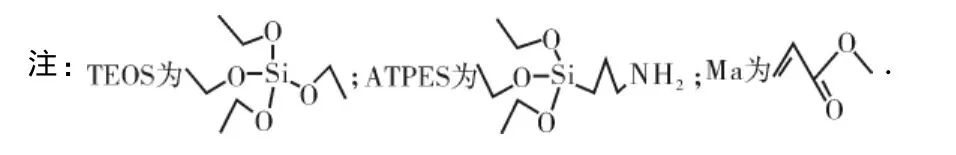
1 实验部分
1.1 试剂与仪器
无水FeCl3,FeSO4·7H2O,氢氧化钠,γ-氨丙基三乙氧基硅烷(APTES),丙烯酸甲酯(MA)、氯化钙、乙二胺(EDA)、乙二胺四乙酸(EDTA),乙醇、氨水质量分数(25%),硝酸均为分析纯,四乙氧基硅烷(TEOS)为化学纯,使用前均未经处理.
X-射线衍射仪(XRD):XD-5A,日本岛津公司生产.FT-IR(用 KBr压片法):Impact 420型,美国尼高力公司生产.热重分析仪(TGA):Q50型,美国生产.扫描电镜(SEM):JSM-5510LV型,日本电子生产.X-射线光电子能谱(XPS):美国PHI-5702/ESCA/SAM 电子能谱仪.原子吸收光谱(AAS):SOLAAR.M6,美国热电公司生产.
1.2 实验方法
1.2.1 四氧化三铁的制备 Fe3O4采用共沉淀法制备,依据的原理为:Fe2++2Fe3++8OH-=Fe3O4+4H2O,采用Fe3+∶Fe2+=2∶1的摩尔比制备.具体步骤如下:在氩气保护下,分别将3.88g无水FeCl3及3.34g FeSO4·7H2O溶于约280mL去离子水中,机械搅拌1h后,滴加含4.32g NaOH的68mL水溶液,直到pH为9,此时溶液中出现大量黑色沉淀,再继续搅拌3h.以磁铁分离,依次用去离子水及乙醇洗至溶液为中性,70℃真空干燥得Fe3O4颗粒.
1.2.2 氨基修饰四氧化三铁的制备(Fe3O4@SiO2—NH2) 氨基功能基的修饰:将上述方法制得的Fe3O4纳米粒子3.25g研磨后分散于150mL乙醇/水(体积比4∶1)溶液中,超声30min后,转入250mL三颈烧瓶中,在氩气保护机械搅拌下,滴加2mL氨水质量分数(25%~28%),1h后,缓慢滴加含4mLTEOS的20mL乙醇溶液,并升温至45℃反应16h.然后再缓慢滴加含8mL APTES的10mL乙醇溶液,反应24h,继而在60℃下反应3h.反应结束后,产物通过磁铁分离,去离子水洗涤后60℃真空干燥.
1.2.3 丙烯酸甲酯及乙二胺修饰四氧化三铁的制备 丙烯酸甲酯及乙二胺修饰四氧化三铁的制备按两步反应获得:一是Michael加成反应,二是羧基与乙二胺(EDA)的酰胺化反应[17].功能基团氨基修饰的磁性纳米颗粒与过量的丙烯酸甲酯在25℃下、甲醇溶剂中反应24h后磁铁分离,乙醇洗涤后,真空干燥得产物G0(Fe3O4@SiO2—NH2@ MA).将G0与过量的乙二胺在25℃下、甲醇溶剂中反应24h,得到悬浊液用磁铁分离,乙醇洗涤多次,真空干燥得产物G1(Fe3O4@SiO2—NH2@MA—EDA).
1.2.4 G0及G1对Ca2+吸附性能测定 配置一定浓度的氯化钙溶液,定容至250mL容量瓶;用EDTA法测得溶液的Ca2+=0.0039mol/L.
将 G0(50mg)加入到40mL 0.0039mol/L Ca2+溶液中,搅拌半小时,借助磁铁分离,用去离子水洗三次,烘干后加入2mL浓度为1∶1(v∶v)的硝酸酸化使其释放出Ca2+,搅拌半小时,静置,磁铁分离,去离子水洗涤,合并洗涤液,并定容至250mL,用原子吸收光谱(SOLAAR.M6)测定Ca2+含量.
G1中Ca2+含量和G0中Ca2+质量含量测定方法相同.

其中,q为吸附容量(mg/g);Cs为样品中钙离子含量(mg/L),Cb为空白样品中钙离子含量(mg/L),V 为溶液体积,m为样品(吸附剂)质量(g).
2 结 果
2.1 纳米颗粒表征及性能
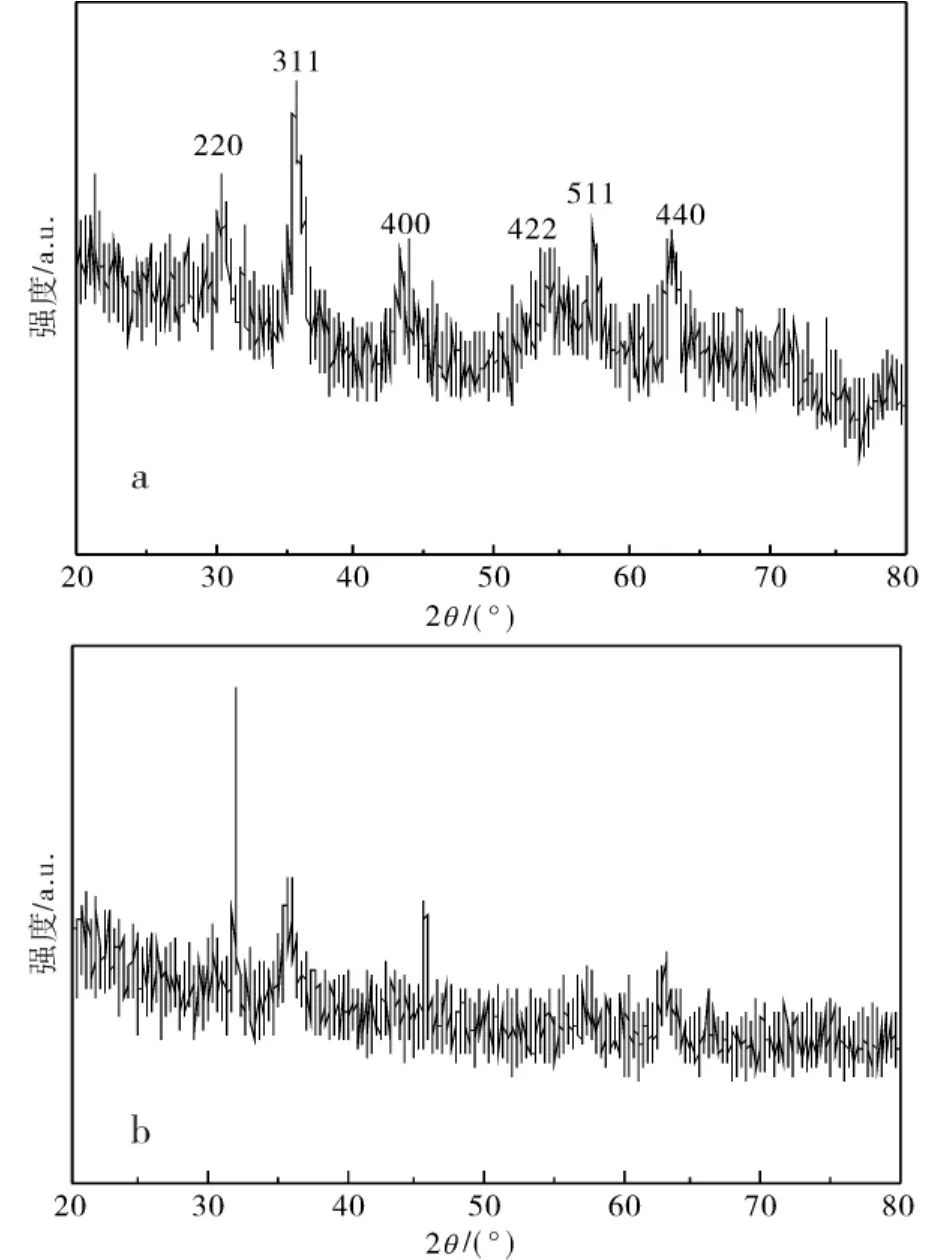
图2 纳米粒子Fe3O4(a)及G1(b)的X-射线衍射图谱Fig.2 XRD patterns of nano-Fe3O4particles(a)and G1(b)
2.1.1 X射线衍射 图2是Fe3O4纳米粒子的X射线粉末衍射图.Fe3O4XRD谱图中,出现了六个衍射峰,分布在2θ=30.3(220),35.5(311),42.9(400),53.3(422),56.6(511),62.2(440)与文献对照,其峰形、相对强度基本吻合[18],对照图a发现,G1的XRD谱图b中,衍射峰强度变弱、变宽,说明有机物的存在[19],按照Scherrer方程[20-21]

计算得到Fe3O4颗粒的平均粒径约为17nm.式中K为Scherrer常数(0.89),λ为X射线波长(0.15406),θ为布拉格衍射角,β为衍射峰的半高峰宽.
2.1.2 扫描电子显微镜 图3是Fe3O4表面修饰功能基团后的扫描电镜图.由图3a可知Fe3O4@SiO2—NH2的微观形貌呈球形,粒径约45nm,存在团聚现象.Fe3O4@SiO2—NH2@MA及Fe3O4@SiO2—NH2@MA—EDA的微观形貌也呈球形,粒径分别约为64和70nm.它们的粒子之间团聚现象较Fe3O4@SiO2-NH2小.这可能是因为在粒子的制备分离过程中采用磁场分离,使得粒子产生一定程度的磁化,造成粒子团聚[22-23].由于后面两个产物的 Fe3O4核的含量相对减少,故由于磁化作用而产生的团聚现象较Fe3O4@SiO2-NH2弱,团聚现象减小.另外,相对于Fe3O4@SiO2-NH2,Fe3O4@SiO2-NH2@MA及Fe3O4@SiO2-NH2@MA-EDA的颗粒尺寸依次增加了19nm和25nm说明已按合成路线图成功接枝.
2.1.3 傅立叶红外表征 采用傅立叶红外表征图谱 (FTIR)对产物进行了表征,结果见图4.图中四个样品均含有中两个较强的吸收峰589、632cm-1,它们可归属于 Fe3O4中 Fe—O 在570cm-1处的分裂所产生的特征伸缩振动峰[24].800cm-1吸收峰是Fe3O4表面—OH伸缩振动峰,说明四个样品中均含有Fe3O4.除样品Fe3O4外,其余三个样品中1069cm-1处的吸收峰为Si—O—Si的伸缩振动吸收峰,表明四氧化三铁颗粒表面成功包裹上一层改性硅胶,2930cm-1和2849cm-1处为C—H的伸缩振动峰,1625cm-1处为N—H的伸缩峰,NH2的弯曲振动峰,3424cm-1处有N—H的伸缩振动峰,1732cm-1处为CO的特征峰,其中样品Fe3O4@SiO2—NH2@MA 在1732cm-1处的峰较 Fe3O4@SiO2—NH2@ MA—EDA强很多,这是由于Fe3O4@SiO2—NH2@MA酰胺化后CO键含量相对减少造成的,这一现象与文献报道的结果类似[25].

图3 Fe3O4@SiO2—NH2(a),Fe3O4@SiO2—NH2@MA(b)及Fe3O4@SiO2—NH2@MA—EDA(c)各产物的扫描电镜图Fig.3 The SEM image of several produds Fe3O4@SiO2—NH2(a),Fe3O4@SiO2—NH2@MA (b)and Fe3O4@SiO2—NH2@MA—EDA(c)

图4 傅立叶红外表征谱图Fig.4 Fe3O4(a),Fe3O4@SiO2—NH2(b),Fe3O4@SiO2—NH2@MA (c)and Fe3O4@SiO2—NH2@MA—EDA(d)的FTIR spectra of Fe3O4(a),Fe3O4@SiO2—NH2(b),Fe3O4@SiO2—NH2@MA (c)and Fe3O4@SiO2—NH2@MA—EDA(d)

图5 样品的热重分析图谱Fig.5 Thermogravimetric analysis curves of Sample
2.1.4 热重分析 由图5知,所有样品均随温度升高而呈现失重现象.可将失重划分为三个部分.25~150℃的失重可归属于物理水分子的逸出;150~800℃的质量损失与有机物的分解逸出及硅醇键的缩合脱水有关.质量损失随着包覆的有机物的增加而增大,这表明Fe3O4颗粒已按合成路线所示的结构逐次包覆了SiO2及相应的有机物.按照图示的质量损失,样品b,c及d中均含有Fe3O4@SiO2—NH2,在温度升到800℃时,所剩余的物质均由Fe3O4及SiO2产生,由于c,d两样品均由b样品依次包覆有机物得到,故除去物理水的失重外(150℃以下),由Fe3O4@SiO2—NH2在150℃至800℃产生的质量损失(Q1)与剩余的物质的质量(m1)之比可近似看作恒定,该值可由b样品算出.由图可知样品c和d在150℃至800℃,Fe3O4@SiO2—NH2所损失的质量(Qn),进而从最后剩余的物质的总质量扣除因组成Fe3O4@SiO2—NH2而损失的质量,可得外层包覆的有机物的质量含量(Org%).计算公式如下:

上式中,Org%表示Fe3O4@SiO2—NH2外包覆的有机物的质量分数;Qn为样品在150~800℃范围内所损失的质量,mn为剩余的物质的质量.按此式计算可得c及d样品中MA及MA—EDA的质量分数分别为6.23%和11.19%.因此TGA也得到了与合成路线吻合的结果.
2.1.5 X射线光电子能谱分析 样品Fe3O4@SiO2—NH2@MA—EDA与钙离子作用前后的XPS全扫描图谱及氧、钙原子的XPS图谱见图6,相应的XPS数据见表1.由Fe3O4@SiO2—NH2@MA—EDA的XPS图中各峰对应原子可知,各元素质量分数(%):C 32.02,O 44.06,Fe 0.69,Si 18.18,N 0.83,这与该样品的设计结构相符,表面所含元素的比例大小依次为O>C>Si>N>Fe,这是由于XPS检测到的是颗粒表面约10nm的结构信息,Fe3O4被包覆在样品颗粒的中心,所以被检测到的铁元素含量最低,这种极少量的铁的检出是由于处于内层的极少数的Fe2p电子非弹性散射产生的.这也说明Fe3O4被硅胶及有机物所包覆,这一现象与文献报道一致[26].Fe3O4@SiO2—NH2@MA—EDA—Ca的图谱中发现,除含有与Fe3O4@SiO2—NH2@MA—EDA相同的原子外,在结合能为346.9eV处出现了一个吸收峰,它可归属于Ca2p,其检测到的含量为1.25%,这与原子吸收光谱结果相符.在CaCl2中Ca2p的电子结合能为347.9eV,而样品中的Ca2p的结合能为346.9降低了1.0eV,表明其电子密度增加,这是由于钙离子与样品中的原子发生了配位作用而形成的[27].
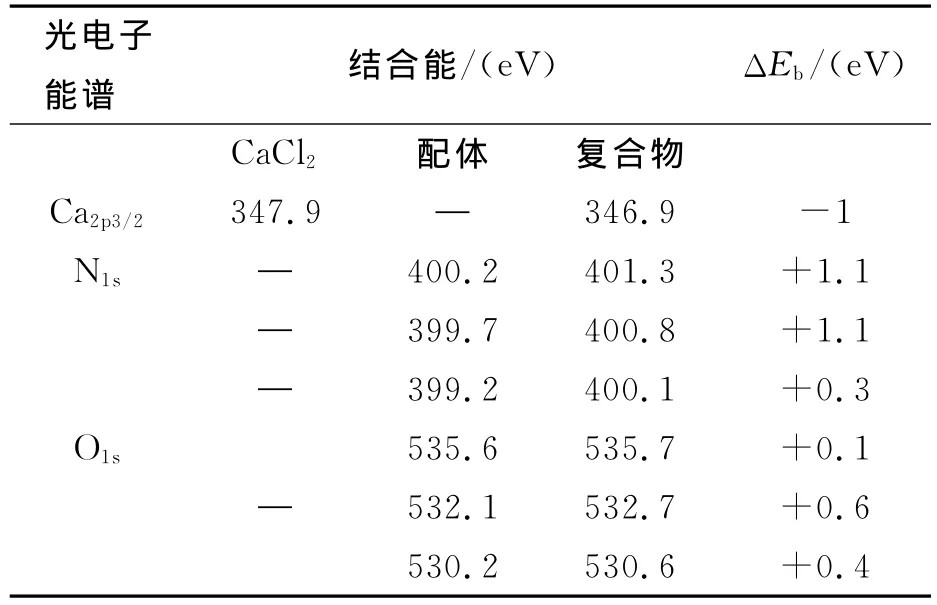
表1 配体(Fe3O4@SiO2—NH2@MA—EDA)及配合物(Fe3O4@SiO2—NH2@MA—EDA—Ca)的XPS数据Table 1 The data of ligand(Fe3O4@SiO2—NH2@MA—EDA)and complex(Fe3O4@SiO2—NH2@MA—EDA—Ca)
Fe3O4@SiO2—NH2@MA—EDA的O1s峰可分为C—O—C,CONH2和C—O—H,螯合钙离子后,结合能分别升高0.1,0.6,0.4,配体及配合物中的 N1s峰可划分为氨基、酰胺基及叔胺,配合物的N1s结合能较配体分别增加了1.1,1.1,0.3eV,这表明配位原子的电子进入了钙离子的轨道而形成了配位键.
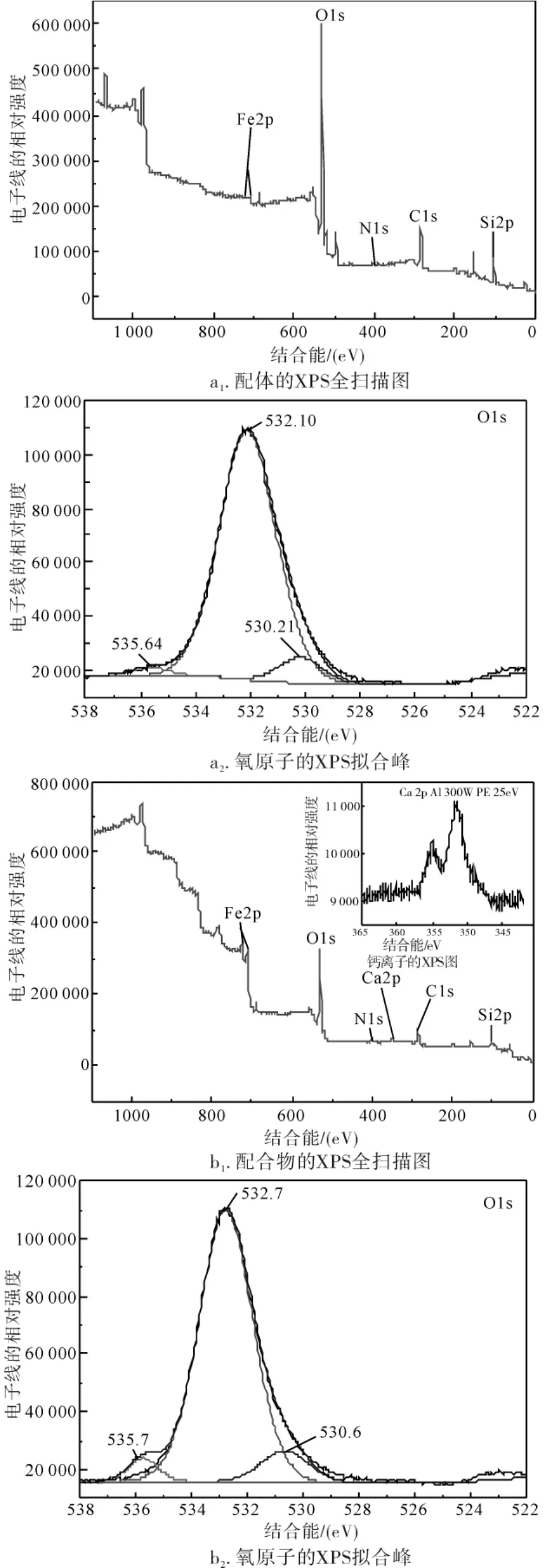
图6 配体(a)和配合物(b)的XPS全扫描图及氧原子的XPS图Fig.6 The XPS scanning pattern of Ligand(a),Complex(b)and atom of oxygen and illustrations for calcium XPS diagram
2.1.6 Fe3O4@SiO2—NH2@MA—EDA与钙离子作用研究[28-29]按前述实验及计算方法得到Fe3O4@SiO2—NH2@MA—EDA 及 Fe3O4@SiO2—NH2@MA—EDA样品中钙含量为分别为0.0108g/g及0.0114g/g.转化为百分含量近似为1.08%和1.14%.其数据及XPS所测得的钙的含量1.25很接近,且与TGA中计算出的MA及MA—EDA相对含量相吻合.本实验结果表明,Fe3O4@SiO2—NH2@MA 及 Fe3O4@SiO2—NH2@MA—EDA样品均可与钙离子结合,但由于所含配位位点较少,一个单位的MA或MA—EDA按配位数最多可结合一个钙离子,故所得的两个样品的钙离子含量均不高.但可以推测采用丙烯酸甲酯及乙二胺进行多步的Michael加成反应及酰胺化反应将逐步提高样品中钙的含量.
3 结 语
a.本文采用共沉淀法制备了两种分别含氨基、酯基及氨基、酰氨基的核壳磁性纳米颗粒,并用SEM,XRD,FTIR,TGA,XPS和TGA进行了表征.
b.磁性纳米颗粒的粒径约为17nm,通过溶胶凝胶法引入氨基后颗粒尺寸为45nm,通过迈克尔加成及酰胺化反应制得的纳米颗粒分别为64nm和70nm.磁性纳米颗形貌规整,为球形.
c.两个样品中MA及MA—EDA的含量分别为6.23%和11.19%,表明样品中MA中的酯基已被乙二胺酰胺化.
d.XPS及与钙离子螯合实验研究表明,钙离子与修饰后的磁性纳米颗粒间以配位键相结合,叔氨基或伯氨基与两个MA或MA—EDA形成了配位空穴可结合一个钙离子,随着接枝的树形分子的代数的增加,吸附钙离子的能力会逐步提高.因此,笔者认为合成多代数的该类磁性纳米颗粒有望用于水中钙离子的吸附.
[1]张杰,胡登华.磁性纳米Fe3O4粒子的制备与应用[J].武汉工程大学学报,2011,33(10):4-8.
[2]李成魁,祁红璋,严彪.磁性纳米四氧化三铁颗粒的化学制备及应用进展[J].上海金属,2009,31(4):54-58.
[3]张书第,张振芳,文松林.化学共沉淀法制备纳米四氧化三铁粉体[J].辽宁化工,2011,40(4):325-327.
[4]Shao Dan Dan,Xu Ke Ke,Song Xiao Jie.Effective adsorption and separation of lysozyme with PAA-modified Fe3O4@silica core/shell microspheres[J].Journal of Colloid and Interface Science,2009,336:526-532.
[5]刘春丽,魏旭.磁性四氧化三铁纳米粒子的合成及改性[J].研发前沿,2009,17(20):20-21.
[6]蒋婷婷,刘喜军,樊珊.磁性高分子微球Fe3O4/PMMA的制备与表征[J].齐齐哈尔大学学报,2012,28(2):53-56.
[7]饶通德.原位聚合法合成Fe3O4/聚丙烯酸纳米粒子及其吸附性能研究[J].西南民族大学学报:自然科学版,2011,37(5):791-794.
[8]王胜碧.含羧基的磁性生高分子微球的制备和表征[J].安顺学院学报,2009,11(6):82-85.
[9]Chih K,Chia H,Chii C.Magnetic SiO2/Fe3O4colloidal crystals[J].Nanotechnology,2008,19:1-5.
[10]王卫伟,姚佳良.柠檬酸盐改性Fe3O4水溶液分散性研究[J].化学研究与应用,2012,24(10):1565-1570.
[11]Yamaura M,Camilo R L,Sampaio L C.Preparation and characterization of (3-aminopropyl)triethoxysilane-coated magnetite nanoparticles[J].Journal of Magnetism and Magnetic Materials,2004,279:210-217.
[12]冯斌,任志强,魏东光.3-氨丙基三乙氧基硅烷表面修饰的磁性Fe3O4纳米粒子合成与表征[J].化工新型材料,2008,36(12):26-29.
[13]郑群雄,刘煌,徐小强,等.羧基化核壳磁性纳米Fe3O4吸附剂的制备及对Cu2+吸附性能[J].高等学校化学学报,2012,33(1):107-113.
[14]Li Cui Lin,Wang Jia qing,Yang Zhi Wang.Baeyer-Villiger oxidation of ketones with hydrogen peroxide catalyzed by cellulose-supported dendritic Sn complexes[J].Catalysis communications,2007(8):1202-1208.
[15]卢康利,陈枫,杨晋涛,等.聚酰胺胺接枝改性纳米二氧化硅及性能研究[J].科技通报,2010,26(5):753-757.
[16]谢英惠,张平,袁俊生.新型分子筛对海水中钙离子的脱除研究[J].非金属矿,2012,35(3):68-70.
[17]Norio T,Hajime I,Toshiya S.Grafting of dendrimerlike'highly branched polymer onto ultrafine silica surface[J].Reactive and Functional Polym,1998,37:75-82.
[18]Yamaura M,Camilo R,Sampaio L.Preparation and characterization of(3-aminopropyl)triethoxysilanecoated magnetite nanoparticles[J].Journal of Magnetism and Magnetic Materials,2004,279:210-217.
[19]Liu Bin,Zhang Wei,Yang Feng Kai.Facile Method for Synthesis of Fe3O4@Polymer Microspheres and Their Application As Magnetic Support for Loading Metal Nanoparticles [J].Journal of Physical Chemistry,2011,115:15875-15884.
[20]Zhao Shen Qiang,Zheng Xin,Li Jie,et al.Synthesis and Graft Polymerization Modification of Fe3O4Magnetic Nanoparticle[J].Materials review,2012,26(2):5-8.
[21]马晓利,尚宏周,赵艳琴,等.纳米四氧化三铁的制备及表面修饰研究[J].精细与专用化学品,2012,20(2):26-31.
[22]Deng Yong Hui,Wang Chang Chun,Shen Xi Zhong.Preparation,Characterization and Application of M-ultistimuli-Responsive Microspheres with Fluorescence -Labeled Magnetic Cores and Thermoresponsive Shells[J].Chemistry A European Journal,2005,11:6006-6013.
[23]张全丽,吴占超,匡少平.羧氨基修饰磁性壳聚糖纳米微球的制备与表征[J].青岛科技大学学报:自然科学版,2011,32(6):573-575.
[24]冯斌,任志强,魏东光.3-氨丙基三乙氧基硅烷表面修饰的磁性Fe3O4纳米粒子合成与表征[J].化工新型材料,2008,36(12):26-29.
[25]肖夏,吴江渝.以二乙醇胺为核聚酰胺胺树形分子的合成及表征[J].武汉工程大学学报,2011,33(11):43-46.
[26]Claire M,Meriem F,Smain B.Magnetic Fe2O3-Polyrene/PPy Core/Shell Particles:Bioreactivity and Self-Assembly [J].Langmuir,2007,23:10940-10949.
[27]苏英草,程贤甦,关怀民.氯化钙一聚乙二醇配位聚合物的配位数研究[J].化学物理学报,1999,12(5):625-632.
[28]吴豫鄂,吴开宇.高磷高钙液体复混肥中钙含量的测定[J].武汉工程大学学报,2009,31(5):17-20.
[29]王轩翾,冉祥兰,刘少文,等.磷石膏中硫酸钙含量测定的方法[J].武汉工程大学学报,2010,32(5):41-44.
