手性二级胺催化的不对称串联反应研究进展
2013-10-22王亮,鲁珍
王 亮,鲁 珍
(江汉大学 光电化学材料与器件省部共建教育部重点实验室,化学与环境工程学院,湖北 武汉 430056)
2000年,List和 Barbas等[1]报道了 l-脯氨酸催化的高选择性、高效率的醛和酮分子间的不对称Aldol反应,并提出了烯胺催化的活化模式。同年,MacMillan等[2]设计了手性咪唑啉酮类二级胺催化剂,并成功地应用于不对称Diels-Alder反应之中,进而发展了亚胺催化的活化模式。这两项开创性的研究报道引起了学者们的广泛关注。至此,有机催化剂发展成为继酶和手性过渡金属催化剂之后的又一类重要的手性催化剂,并且逐渐发展成为一个重要的研究方向。
1 活化模式
相对于金属催化剂,有机催化剂具有如下优势:制备简单,原料来源广泛;毒性低、性质稳定;反应操作简单、环境友好。目前,应用于不对称催化的有机催化剂种类很多,主要类型有手性的胺类催化剂,手性氢键催化剂,手性膦酸催化剂,手性相转移催化剂,手性卡宾催化剂等。其中,手性二级胺就是这类催化剂中研究最活跃、应用最广泛的有机催化剂[3]。
手性二级胺可以通过不同的活化模式来催化各种类型的反应。在烯胺催化的反应过程中,手性二级胺能够与醛或酮等羰基化合物作用形成烯胺中间体,提高底物HOMO轨道的能量,从而使得羰基化合物的α位更具亲核性,提高了醛酮底物的活性。能够与各种的亲电试剂反应,获得各种手性分子。在亚胺催化的反应过程中,手性二级胺与α,β不饱和醛酮等羰基化合物结合形成亚胺离子中间体,降低LUMO轨道的能量,从而羰基化合物的β位更具有亲电性,能够受到各种亲核试剂的进攻,获得各种手性的化合物[4-5]。近年来,随着串联反应的逐渐发展及其相关工作的深入研究,结合手性二级胺催化反应的两种活化模式,逐渐发展了各种新型的串联反应,并且取得了很多重要的进展,已经成为一种构建复杂化合物的关键策略[6-8]。
笔者将按照活化模式分类对近些年手性二级胺催化的不对称有机串联反应作出综述。
2 亚胺-烯胺活化
亚胺-烯胺活化是目前在串联反应中应用最普遍的活化模式之一,在这类反应过程中,手性二级胺首先与不饱和羰基化合物形成亚胺离子中间体,随后与亲核试剂发生Michael加成反应,形成一个手性中心并产生烯胺中间体,形成的烯胺中间体是一个很好的亲核试剂,能够与另一分子的亲电试剂发生加成反应,形成第二个手性中心(图 1)[4-5]。
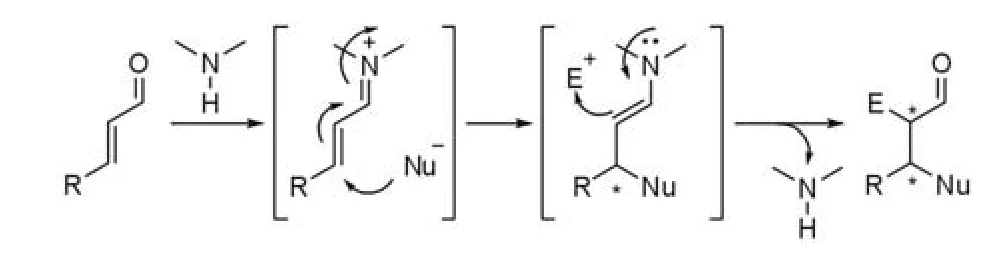
图1 亚胺-烯胺活化模式
2.1 Michael-Aldol反应
2000年,Barbas等[9]报道了l-脯氨酸催化的串联的不对称Michael-Aldol反应(图2)。在这个反应中,l-脯氨酸首先与甲基乙烯基酮1形成亚胺中间体,然后与1,3-环己二酮发生Michael加成反应,形成一个季碳中心。紧接着产生的烯胺中间体与1,3-环己二酮的一个羰基发生Aldol反应,脱去一分子的水,从而一步合成出环状不饱和酮产物4。虽然该反应的收率和对映选择性不高,但是也能够达到用传统的两步法所得的相似结果[2]。

图2 Michael-Aldol反应(Barbas等,2000年)
2004年,Jørgensen[10]等报道了咪唑啉类催化剂7催化的 β-酮酯与α,β-不饱和酮5的串联Michael-Aldol反应(图3)。能够以高收率,高对映选择性地获得具有3个连续手性中心的环己酮类化合物8,该产物经过简单的合成转化后能够获得有用的合成中间体。
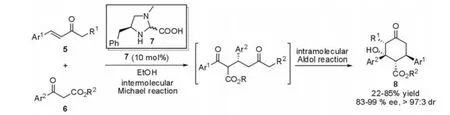
图3 Michael-Aldol反应(Jørgensen等,2004年)
最近,MacMillan等[11]利用这类活化模式成功地实现了烯烃的氢胺化、氢氧化及胺氧化等一系列的串联催化反应,高对映选择性地得到了相应的串联反应产物(图4)。
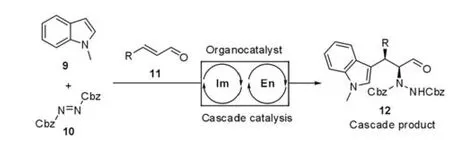
图4 Michael-Mannich反应
2.2 Michael-Aldol-Dehydration反应
2006年,Wang等[12]发展了脯氨醇硅醚15催化的水杨醛(X=O)与α,β-不饱和醛13的对映选择性串联的Michael-Aldol-Dehydration反应。通过合理调控反应条件,在脯氨醇硅醚催化剂的作用下,能够以最高99%的收率以及99%的对映选择性获得串联反应的产物16。该串联反应方法底物使用范围很广(X=S,O,N),能够合成具有重要生理活性的苯并吡喃、苯并硫代吡喃以及喹啉类化合物(图5)。
上述的串联反应过程中,第一步的Michael加成步骤都是由含有杂原子的亲核试剂引起的,如S,O,N等。受到该策略的启发,Enders等[13]于2007年报道了硝基化合物17作为碳亲核试剂参与的串联的Michael-Aldol-Dehydration反应(图6)。在反应过程中,硝基的α-位与α,β-不饱和醛发生Michael加成反应,并形成烯胺中间体,随后与硝基化合物内的羰基发生Aldol反应,最后脱去水形成不饱和的双键。
2.3 Michael-Michael反应
Wang等[14-16]利用具有巯基的 α ,β-不饱和酯22作为底物,发展了新型的脯氨醇硅醚催化下的串联Michael-Michael加成反应(图7)。高选择性的合成具有多手性中心,高官能化的四氢噻吩类衍生物23。这一串联反应策略也为高效快速地合成四氢噻吩类衍生物的提供了一条有效的途径。
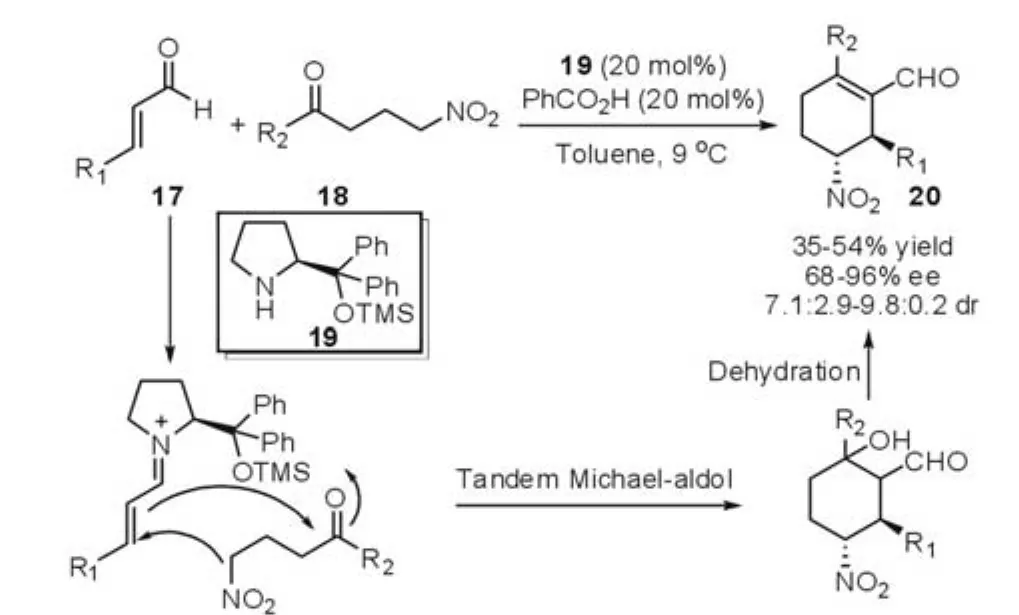
图6 Michael-Aldol-Dehydration反应(Enders等,2007年)
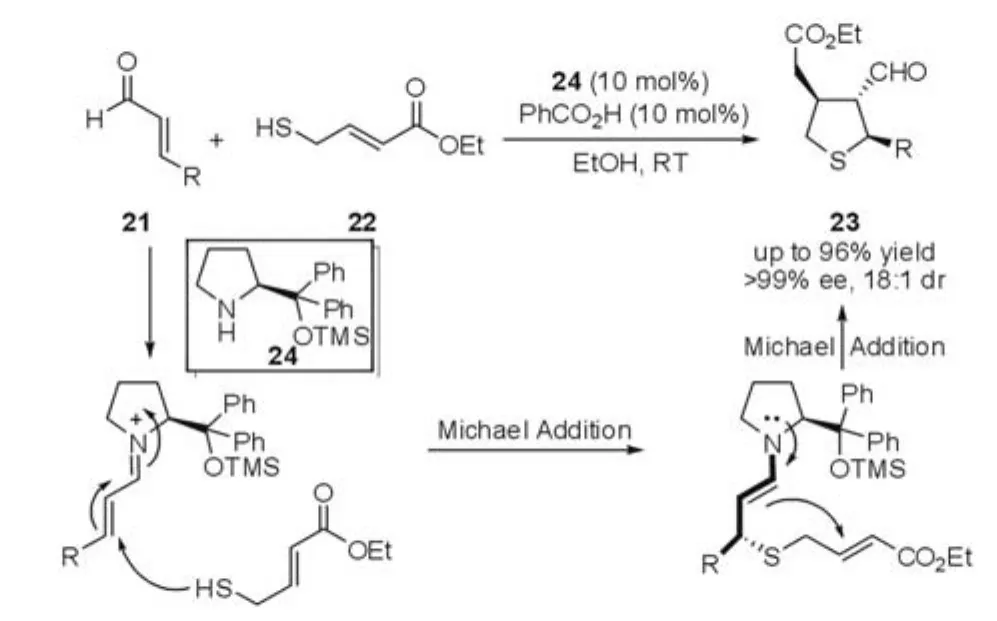
图7 Michael-Michael反应
2.4 Michael-Halogenation反应和氢化-卤代反应
2005年MacMillan等[17]报道了手性咪唑啉酮类催化剂29催化的串联的Michael-Halogenation反应(图8)。通过对各种条件的筛选,当使用化合物27作为氯代试剂时,各种不饱和的醛能够与呋喃、吲哚等亲核试剂顺利发生串联的Mi⁃chael-Halogenation反应,并能够取得最高99%的对映选择性。值得一提的是此反应具有高度的区域选择性,其产物以顺式为主。
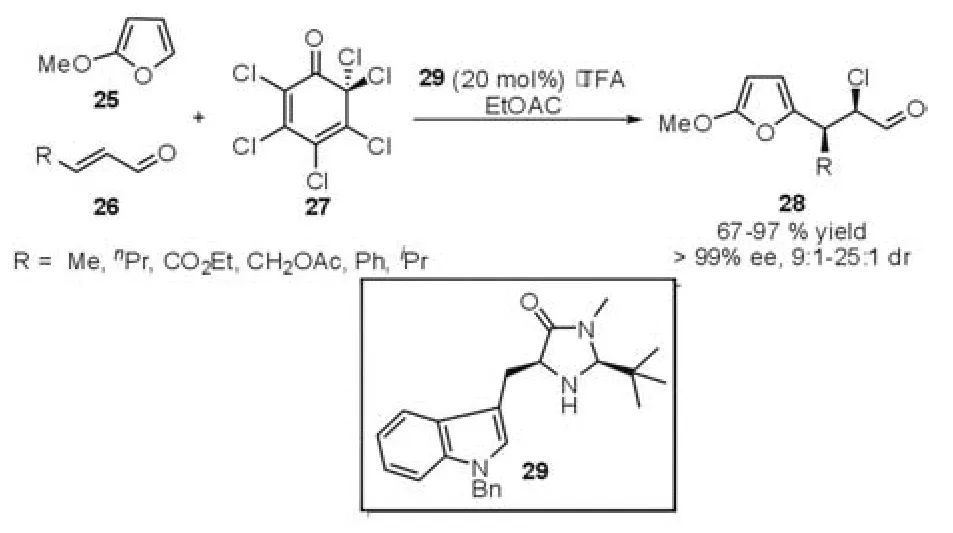
图8 Michael-Halogenation反应
与此同时,作者还成功地将这一策略应用于串联的Hydrogenation-Halogenation反应中。此串联反应是第一例形式上的HCl、HF对三取代的烯烃的不对称加成。进一步地研究还发现一个有趣的现象:当使用氯化物27加成时主要得到是顺式产物,而氟化物33加成则主要得到反式产物。通过调节不同的手性二级胺催化剂,可以有效地控制加成产物的选择性(图9)。
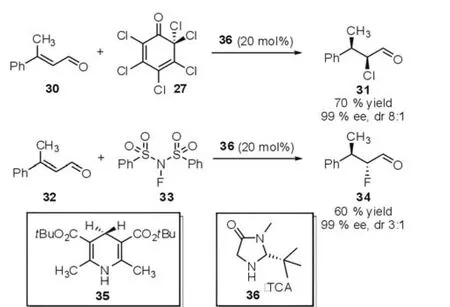
图9 Hydrogenation-Halogenation反应
2.5 Hydrogenation-Michael反应
此外,Yang等[18]小组通过合理的设计底物,在咪唑啉酮催化剂38的作用下,化合物37能够与Hantzsch酯发生不对称串联的Hydrogena⁃tion-Michael环化反应(图10)。作者对反应的条件进行了详细的研究,发现在1,4-二氧六环作为溶剂,使用催化剂38能够获得最好的结果。
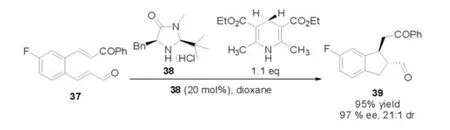
图10 Hydrogenation-Michael反应
2.6 Michael-Alkylation反应
在Kunz等建立的稳定硫叶立德与α,β-不饱和醛反应生成环丙烷的研究基础之上,Wang等[19]于2005年报道了氢化吲哚甲酸42催化的串联的Michael-Alkylation反应(图 11),对反应机理和过渡态进行了详细的研究,发现催化剂的结构改变对产物的对映选择性有重要影响。由于苯环空间位阻作用,以及底物与苯环中的氢原子之间的位阻作用,使催化剂与α,β-不饱和醛形成优势的(Z)式亚胺离子中间体,从而能以较高的对映选择性(89-96%ee)获得多取代、高官能化的环丙烷衍生物43。
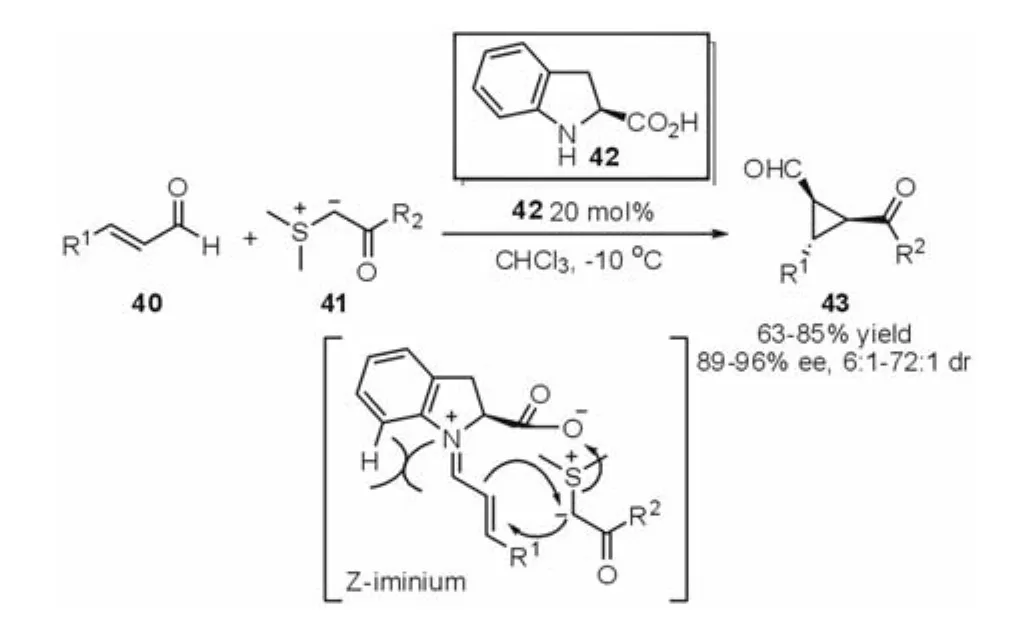
图11 Michael-Alkylation反应(Wang等,2005年)
同年,Jørgensen等[20]报道了首例基于亚胺-烯胺活化模式的环氧化反应(图12)。此反应也可以认为是一个串联的oxo-Michael-Alkylation反应。当以脯氨醇硅醚45作为催化剂,H2O2作为环氧化试剂时,能够得到较高的对映选择性和较好的收率。在此例研究报道之后,文献[21-24]对烯烃环氧化反应进行了报道。

图12 oxo-Michael-Alkylation反应
除了上述的手性二级胺催化串联反应构建环氧化合物之外,2007年,Córdova等[25]报道了脯氨醇硅醚49催化的不饱和醛47与氮化物48发生的aza-Michael-Alkylation反应(图13)。该反应底物使用范围较广,反应条件温和,其产物能够经过简单的两步反应转化为氨基酸酯的衍生物50。
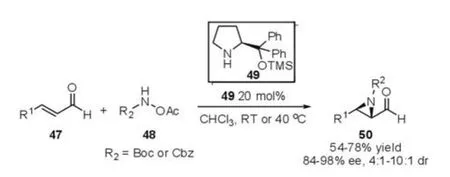
图13 aza-Michael-Alkylation反应
在串联的Michael-烷基化反应过程中,与硫叶立德相比,价廉易得、性质稳定的卤代物更适合作为反应底物。但是,烷基卤代物在反应过程中可能直接与催化剂反应,从而使催化剂失去催化效果。Wang等[26]发展了脯氨醇硅醚催化的不饱和醛与溴代的丙二酸酯53发生的新型串联的Michael-烷基化反应。通过改变反应的条件,当使用2,6-二甲基吡啶时,反应能够高选择性的获得多取代的环丙烷类化合物55(图14)。
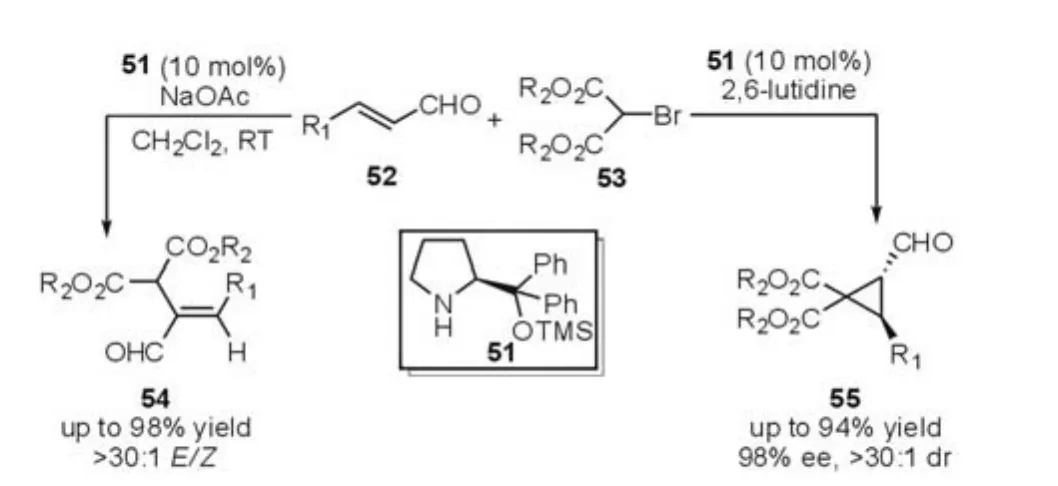
图14 Michael-Alkylation反应(Wang等,2007年)
采用Michael-Alkylation反应能够高效构建多取代三元环化合物,并且通过此策略还可以用于构建五元环状化合物。例如,Córdova等[27]于2007年发展了脯氨醇硅醚催化的溴代乙酰乙酸乙酯57与不饱和醛的串联Michael-Alkylation反应,能够高对映选择性地合成多取代的环戊烷类衍生物59(图 15)。
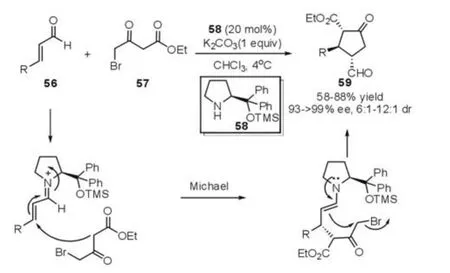
图15 Michael-Alkylation反应(Córdova等,2007年)
2.7 Michael-Amination反应
2005年,Jørgensen等[28]报道了第一例多组分的硫杂的Michael加成/胺化串联反应(图16)。在反应过程中,催化剂首先与α,β-不饱和醛形成亚胺,然后与底物60进行Michael加成,随后生成的烯胺再与偶氮化合物61进行加成反应,最终高对映选择性(97-99%ee)地获得串联反应的产物(图16)。其产物非常有用,经过硼氢化钠还原和酯交换能够顺利地获得噁唑啉酮类衍生物。值得注意的是,反应温度对该反应的对映选择性和反应速率有很大的影响:在室温下,第一步硫杂的Michael加成能很快地完成,但是加成产物容易消旋化;当温度降至-15℃时,虽然能很好地抑制Michael产物的消旋化,但反应速率变慢。最后,作者发现向反应体系中加入苯甲酸作为共催化剂,能够大大提高了反应速率。
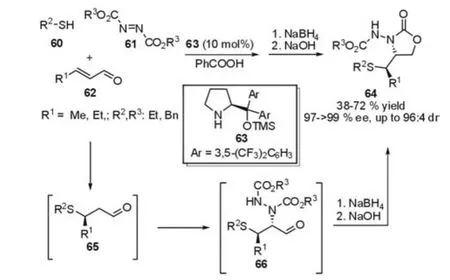
图16 Michael-Amination反应
3 烯胺-亚胺活化
烯胺-亚胺活化模式原理上是亚胺-烯胺活化模式的逆转过程。在烯胺-亚胺活化模式中,首先催化剂与底物形成烯胺中间体,与亲电试剂作用后,烯胺中间体会转化为亚胺中间体,随后生成的亚胺再与另一分子亲核试剂反应,就得到了最终的多取代的目标产物。
3.1 Mannich-aza-Michael反应
2003年,Ohsawa等[29]发展了l-脯氨酸催化的串联的Mannich-aza-Michael反应来构建天然的吲哚生物碱片段(图17)。作者对反应机理进行了研究,发现此过程类似于形式上的aza-Diels-Alder反应。首先l-脯氨酸与甲基乙烯基酮68作用形成烯胺中间体,与吲哚底物67中亚胺官能团发生加成反应形成了亚胺中间体71。然后发生分子内的aza-Michael加成反应形成产物。
在此工作基础之上,2006年,Ohsawa等[30]将它们发展起来的这种策略成功地应用于天然产物ent-dihydrocorynantheol的不对称合成之中,并取得了很好的对映选择性(99%ee)和非对映选择性(>99:1 dr,图 18)。
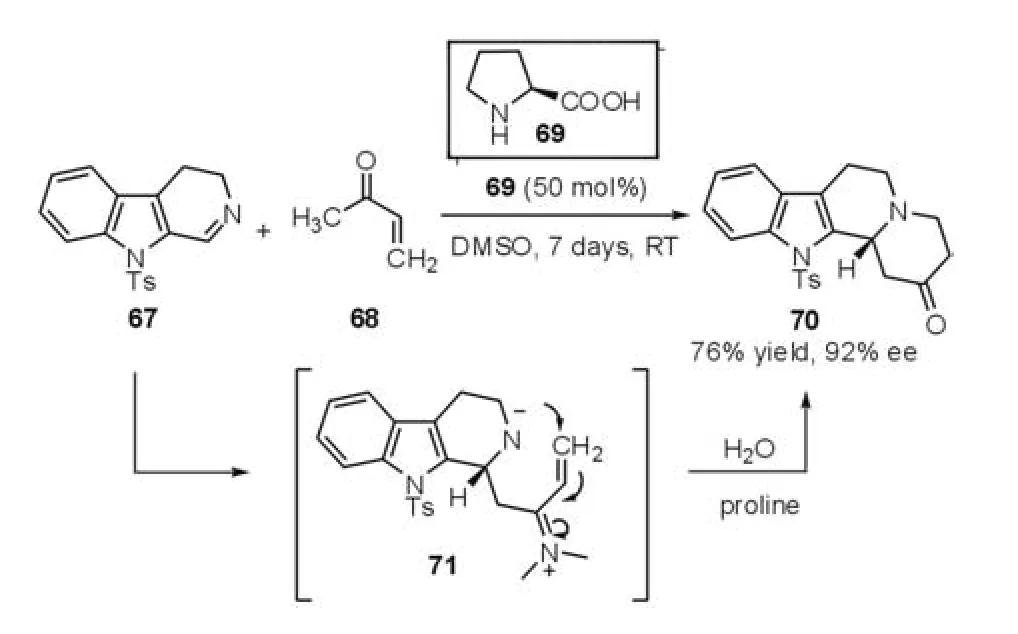
图17 Mannich-aza-Michael反应(Ohsawa等,2003年)
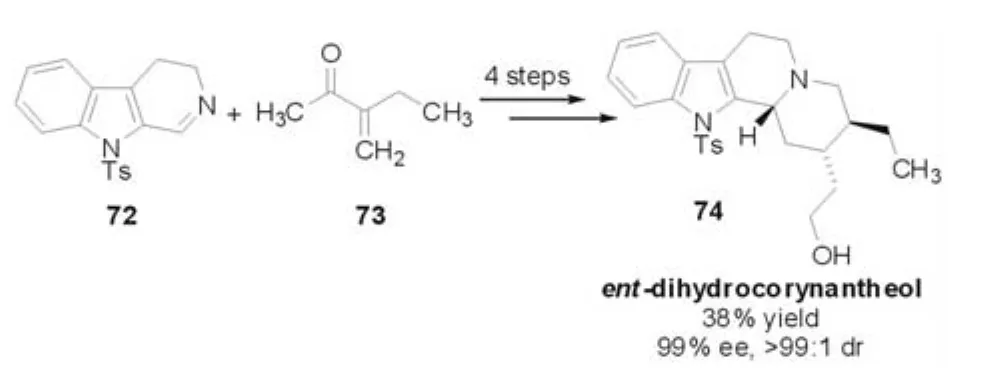
图18 Mannich-aza-Michael反应(Ohsawa等,2006年)
2005年,Córdova等[31]报道了 l-脯氨酸催化的三组分的串联的Mannich-aza-Michael反应(图19)。在反应过程中,l-脯氨酸与底物75形成二烯体,甲醛与苯胺77形成亚胺,然后二烯体与原位产生的亚胺发生形式上的aza-Diels-Alder反应,最终得到桥联杂环类化合物。研究发现,在室温至50℃范围内,以DMSO作为溶剂,可以高收率、高对映选择性地获得目标产物(94-99%ee)。
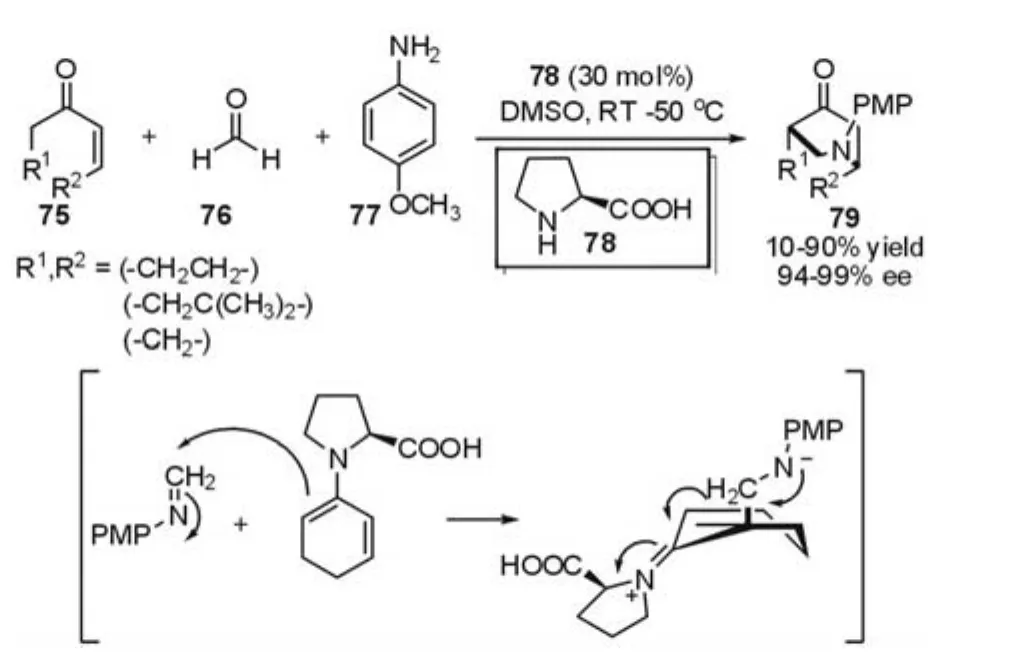
图19 Mannich-aza-Michael反应(Córdova等,2005年)
3.2 Aminooxylation-Michael反应
Yamamoto等[32]结合有机催化的α-aminooxyl⁃ation反应和Michael加成反应的特点,利用吡咯烷衍生物82催化了亚硝基苯与环己烯酮发生的串联的亚硝基Aminooxylation-Michael反应(图20),能够高效、高对映选择性的合成桥联的新型六元杂环化合物83。研究发现,该反应是一个形式上的Diels-Alder反应,并且在实际反应过程中吡咯烷衍生物催化剂比常见的l-脯氨酸催化剂效果要好。
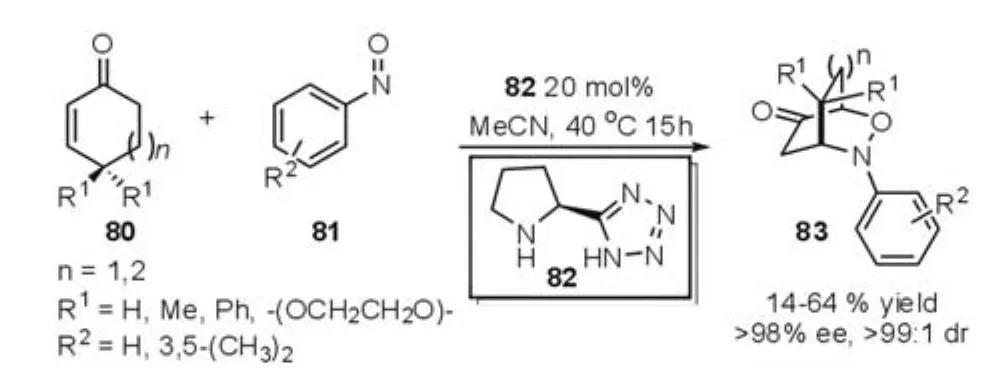
图20 Aminooxylation-Michael反应
4 结语
利用手性二级胺催化的不对称串联反应已经迅速发展为有机合成中一种高效且强有力的工具,根据不同的催化活化模式可以设计一系列新颖的串联反应过程,从而得到各种具有复杂结构的目标分子。研究者将不断寻求使用更加简捷的操作过程和温和的反应条件来发展各类新型的有机催化的串联反应,并且,人们开始逐渐认识到使用有机催化的串联反应来构建复杂的分子骨架已经不再是传统的自然界酶的催化领域。在未来,有机催化的串联反应的发展方向将是通过选择新的活化模式、设计新的底物来持续地扩展这类反应的应用范围,并将其应用于具有生物活性分子及天然产物的合成中。
[1]List B,Lerner R A,Barbas C F.Proline-catalyzed di⁃rect asymmetric aldol reactions[J].J Am Chem Soc,2000,122:2395-2396.
[2]Ahrendt K A,Borths C J,MacMillan D W C.New strat⁃egies for organic catalysis:the first highly enantioselec⁃tive organocatalytic diels-alder reaction[J].J Am Chem Soc,2000,122:4243-4244.
[3]Erkkilä A,Majander I,Pihko P M.Iminium catalysis[J].Chem Rev,2007,107:5416-5470.
[4]List B,Yang J W.The organic approach to asymmetric catalysis[J].Science,2006,313:1584-1586.
[5]MacMillan D W C.The advent and development of or⁃ganocatalysis[J].Nature,2008,455:304-308.
[6]Wasike J C,Obrey S J,Bazan G C.Concurrent tandem catalysis[J].Chem Rev,2005,105:1001-1020.
[7]Nicolaou K G,Edmonds D J,Bulger P G.Cascade re⁃actions in total synthesis[J].Angew Chem Int Ed,2006,45:7134-7186.
[8]Grondal C,Jeanty M,Enders D.Organocatalytic cas⁃cade reactions as a new tool in total synthesis[J].Na⁃ture Chem,2010(2):167-178.
[9]Bui T,Barbas C F.A proline-catalyzed asymmetric Co⁃binson annulation reaction [J].Tetrahedron Lett,2000,41:6951-6954.
[10]Halland N,Aburel P S,Jørgensen K A.Highly enan⁃tio-and diastereoselective organocatalytic asymmetric domino michael–aldol reaction of β-ketoesters and α,β-unsaturated ketones[J].Angew Chem Int Ed,2004,43:1272-1277.
[11]Simmons B,MacMillan D W C.Cycle-specific organo⁃cascade catalysis:application to olefin hydroamina⁃tion,hydro-oxidation,and amino-oxidation,and to natural product synthesis[J].Angew Chem IntEd,2009,48:4349-4353.
[12]Wang W,Li H,Zu L S.Enantioselective organocatalyt⁃ic tandem michael-aldol reactions:one-pot synthesis of chiral thiochromenes[J].J Am Chem Soc,2006,128:10354-10355.
[13]Enders D,Narine A A,Benninghaus T.Asymmetric or⁃ganocatalytic domino reactions of γ-Nitroketones and enals[J].Synlett,2007,11:1667-1670.
[14]Wang J,Li H,Wang W.Organocatalytic enantioselec⁃tive cascade michael–aldol condensation reactions:ef⁃ficient assembly of densely functionalized chiral cyclo⁃pentenes [J].Angew Chem IntEd,2007,46:9050-9053.
[15]Zu L S,Li H,Wang W.Synthesis of highly functional⁃ized chiral cyclopentanes by catalytic enantio-and dia⁃stereoselective double michael addition reactions[J].Angew Chem Int Ed,2007,46(20):3732-3734.
[16]Li H,Zu L S,Wang W.Enantioselective epoxidation of α,β-Enones promoted by α,α-Diphenyl-l-proli⁃nol as bifunctional organocatalyst[J].Org Lett,2007(9):1833-1836.
[17]Huang Y,Walji A M,MacMillan D W C.Enantioselec⁃tive organo-cascade catalysis[J].J Am Chem Soc,2005,127:15051-15052.
[18]Yang J W,Hechavarria F M T,List B.Catalytic asym⁃metric reductive michael cyclization[J].J Am Chem Soc,2005,127:15036-15037.
[19]Kunz R K,MacMillan D W C.Enantioselective organo⁃catalytic cyclopropanations:The Identification of a New Class of Iminium Catalyst Based upon Directed Electro⁃static Activation[J].J Am Chem Soc,2005,127:3240-3241.
[20]Marigo M,Franzen J,Jørgensen K A.Asymmetric or⁃ganocatalytic epoxidation of α,β-Unsaturated alde⁃hydes with hydrogen peroxide[J].J Am Chem Soc,2005,127:6964-6965.
[21]Lee S,MacMillan D W C.Enantioselective organocata⁃lytic epoxidation using hypervalent iodine reagents[J].Tetrahedron,2006,62:11413-11424.
[22]Sundèn H,Ibrahem I,Córdova A.Direct organocatalyt⁃ic asymmetric epoxidation of α,β-Unsaturated alde⁃hydes[J].Tetrahedron Lett,2006,47:99-103.
[23]Lattanzi A. Enantioselective epoxidation of α,β-Enones promoted by α,α-Diphenyl-l-prolinol as bifunctional organocatalyst[J].Org Lett,2005(7):2579-2580.
[24]Li Y,Liu X,Zhao G.4-Substituted-α,α-diaryl-proli⁃nols improve the enantioselective catalytic epoxidation of α,β-enones[J].J Org Chem,2007,72:288-291.
[25]Vesely J,Ibrahem I,Córdova A.Organocatalytic enan⁃tioselective aziridination of α,β-Unsaturated aldehydes[J].Angew Chem Int Ed,2007,46:778-781.
[26]Xie H X,Zu L S,Wang W.Organocatalytic enantiose⁃lective cascade michael-alkylation reactions:synthesis of chiral cyclopropanes and investigation of unexpected organocatalyzed stereoselective ring opening of cyclopro⁃panes[J].J Am Chem Soc,2007,129:10886-10887.
[27]Rios R,Vesely J,Córdova A.One-pot organocatalytic domino michael/α-alkylation reactions:highly enanti⁃oselective synthesis of functionalized cyclopentanones and cyclopentanols[J].Tetrahedron Lett,2007,48:5835-5839.
[28]Marigo M,Schulte T,Jørgensen K A.Asymmetric multi⁃component domino reactions and highly enantioselec⁃tive conjugated addition of thiols to α,β-unsaturated al⁃dehydes[J].J Am Chem Soc,2005,127:15710-15711.
[29]Itoh T,Yokoya M,Ohsawa A.Proline-catalyzed asym⁃metric addition reaction of 9-tosyl-3,4-dihydro-βcarboline with ketones[J].Org Lett, 2003(5):4301-4302.
[30]Itoh T,Yokoya M,Ohsawa A.Total synthesis of ent-di⁃hydrocorynantheol by using a proline-catalyzed asym⁃metric addition reaction[J].Org Lett, 2006, 8:1533-1534.
[31]SundQn H,Ibrahem I,Eriksen L,et al.Direct catalyt⁃ic enantioselective aza-diels– alder reactions[J].An⁃gew Chem Int Ed,2005,44:4877-4880.
[32]Yamamoto Y,Momiyama N,Yamamoto H.Enantiose⁃lective tandem o-nitroso aldol/michael reaction[J].J Am Chem Soc,2004,126:5962-5963.
