草酸根及对咪唑基苯甲酸根构筑的三重互穿镉ギ配合聚合物的合成、结构与发光性能研究
2013-10-17张艳萍王莉丽刘连利田爱香
张艳萍 王莉丽 刘连利 田爱香 应 俊
(渤海大学实验管理中心,锦州 121000)
具有穿插结构的配位聚合物因其在磁学、光学、电学、催化、气体存储与吸附等领域的潜在应用,而吸引了大量研究者的热情,其性质和结构之间的关系更是人们研究与关注的热点[1-7]。尽管穿插的影响因素没有最终确定,但是通过对现有穿插结构配合物的研究,研究者通过改变有机桥联配体的长度、配体的配位元素,以及配位模式,或者使用混合配体来构筑穿插结构的方式[8-12]获得结构新颖的性质优异的配位化合物,从而进一步对配合物结构与性质的关系进行总结[13-14]。草酸根作为桥联配体时[15],配位模式多样,能够方便地改变耦合中心的配位环境,具有较强的传递电子能力;而咪唑羧酸及其衍生物[16-18]不但具有羧酸类配体的配位特点,并且其中的咪唑基团含有π电子共轭体系。因此,利用草酸根和咪唑羧酸类配体共同构筑具有新奇结构的配位聚合物成为一个热点方向。目前以对咪唑基苯甲酸(HL)作为有机组分的配合物报道较少[19]。本文通过水热法,利用草酸与对咪唑基苯甲酸为有机组分,Cd(NO3)2·2H2O为无机原料,合成了一个具有三重互穿网络的镉配位聚合物:[Cd(L)(C2O4)0.5(H2O)]n,测定了该化合物的晶体结构,并研究了其光致发光性质。
1 实验部分
1.1 仪器和试剂
Cd(NO3)2·2H2O,草酸和对咪唑基苯甲酸均为国产分析纯试剂。红外光谱(IR)采用美国Nicolet公司Alpha-Centauri 560型FT-IR光谱仪,样品经溴化钾压片,扫描范围4 000~400 cm-1;晶体结构用德国布鲁克公司CCD面探X-射线衍射仪测定。样品荧光光谱用Hitachi F-4500光谱仪测定;紫外光谱用UV-2550光谱仪测定;X-射线粉末衍射数据用理学Ultima IV 衍射仪上收集,Cu Kα(λ=0.154 18 nm)射线;热重曲线用美国PE公司Pyris Dianond TG/DTA热分析仪测定。
1.2 标题化合物的合成
将对咪唑基苯甲酸(0.1 g),Cd(NO3)2·2H2O(0.20 g),草酸(0.03 g)和 10 mL H2O 混合,在搅拌条件下以1.0 mol·L-1HNO3调节体系 pH 值至 4.0。 室温下剧烈搅拌1 h,然后将反应混合物置于20 mL内衬聚四氟乙烯的不锈钢反应釜中,放入120℃烘箱内,在自生压力条件下反应3 d后逐渐冷却到室温,经过滤、去离子水洗涤后于室温下晾干,得到绿色块状晶体,产率(以C元素计):15%。C11H9CdN2O5的元素分析结果(括号内为理论值,%):C 37.42(37.54),H 2.49(2.51),N 5.56(5.59)。
1.3 晶体结构的测定
选取大小为 0.22 mm×0.12 mm×0.08 mm 的蓝色单晶样品粘在玻璃纤维上,置于Buker SMART-1000 CCD面探X射线单晶衍射仪进行数据收集,用 Mo Kα 射线(λ=0.071 073 nm),以 ω 扫描方式在293K下收集衍射数据。所有数据经LP因子校正,用直接法得到全部非氢原子坐标(SHELXS-97 Sheldrick,1990),有机基团上的氢原子坐标采用几何加氢的方法得到,用全矩阵最小二乘法精修结构(SHELXL-97 Sheldrick,1997)[20]。晶体学数据见表1,选择的部分键长和键角见表2。
CCDC:874995。
2 结果与讨论
2.1 红外光谱
标题配合物的红外光谱是以KBr为基质,在4 000~400 cm-1范围内测定的。根据文献[21]对红外光谱中主要吸收峰进行归属:在配合物的红外光谱中可以看出,1 243和721 cm-1的吸收峰对应于咪唑环的伸缩振动;而1 391和1 680 cm-1的吸收峰则归属于羧酸中的羧基的伸缩振动;在3 500~3 000 cm-1区域的宽吸收峰归属为形成氢键的水分子的伸缩振动吸收峰。
2.2 标题化合物的晶体结构
标题化合物的晶体学数据与主要的键长、键角列于表1和表2。X-射线晶体学研究表明,化合物1分子不对称结构单元包含1个镉离子,0.5个草酸分子,1个L配体分子和1个配位水分子(图1)。该化合物只包含一个晶体学独立的六配位的Cdギ,连接一个草酸分子的2个氧原子(O3i,O4i),L配体的2个羧基氧原子(O1,O2)和一个氮原子(N1),一个配位水分子(O1W)。每一个Cdギ均采取畸变八面体配位构型,Cd-O 键长范围为 0.224 2~0.239 0 nm,Cd-N 的键长均为0.225 1 nm。其中,Cd-O键最短的是配位水上的O与Cd的距离0.224 2 nm,明显短于其他的Cd-O键,因此水中氧原子的配位能力最强。

表1 标题化合物的晶体学参数Table1 Crystal data and structure refinements for title complex
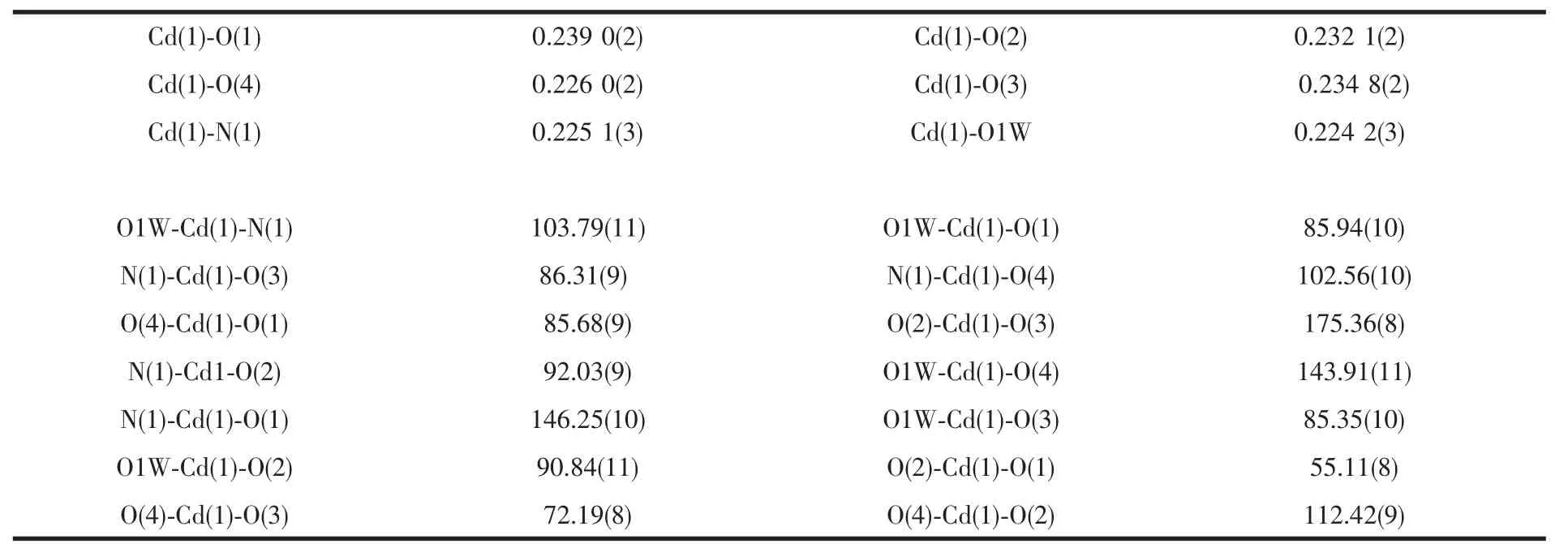
表2 标题化合物选择的键长与键角Table 2 Selected bond lengths(nm)and bond angles(°)for title complex
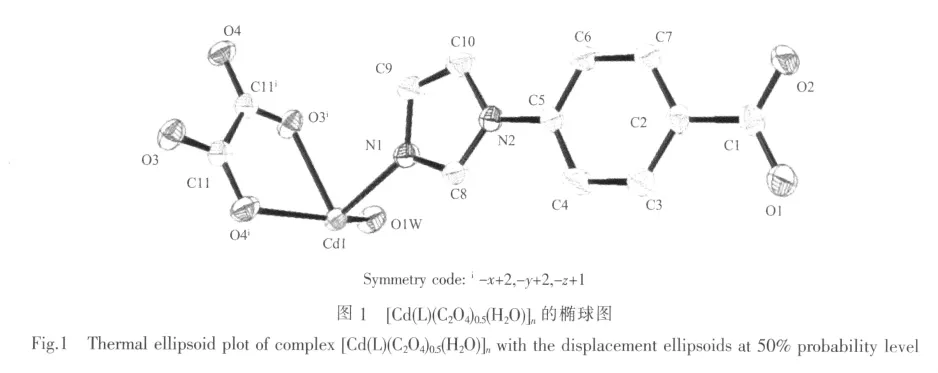
混合配体L和草酸发挥了桥连作用连接Cdギ离子构筑整体框架,并且所有的配位点都得到了充分的利用。其中L配体的1个N原子和2个羧基氧原子分别连接2个Cdギ,草酸分子的四个氧原子也与2个Cdギ进行配位从而形成了Cdギ单元。配体L将Cdギ单元桥连起来,构成二维层状结构,如图2(a)。该二维层中包含尺寸较大的菱形格子,尺寸为1.25 nm×1.50 nm,由4个L配体和4个双核Cdギ单元构成。由于该菱形格子尺寸较大,可能导致化合物的结构不稳定。
因此,3组完全相同的二维层状结构相互穿插从而形成了三重互穿的框架如图2(b)。该三重互穿网络在配合物领域并不太常见,并且三重互穿网络之间通过氢键作用形成三维超分子结构。
2.3 化合物的紫外吸收及荧光光谱
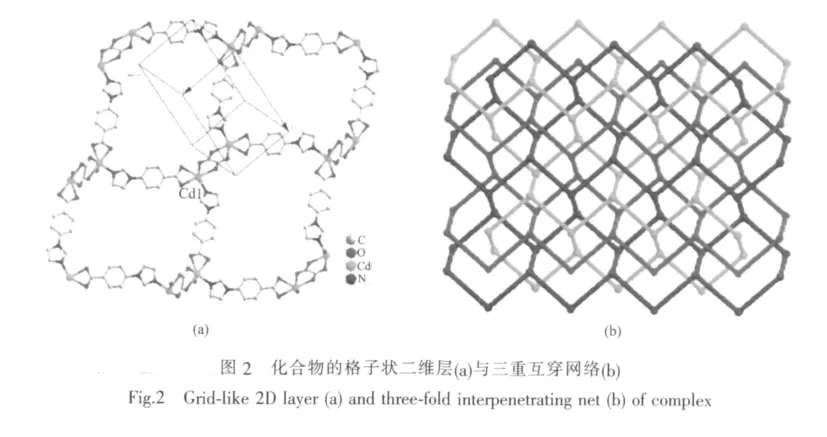
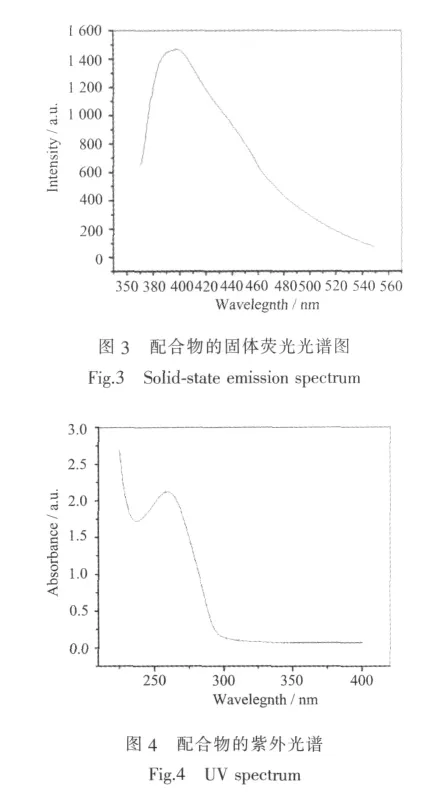
配合物与配体在室温下进行了固体荧光的测试,其中配合物的荧光光谱如图3所示。由于配合物与配体的发射行为很相似,所以我们认为该荧光发射是配体分子内π-π电子跃迁导致的,通过配合物的紫外吸收光谱也得到了证实。配合物在260 nm处有吸收峰,是由配体分子内π-π电子跃迁所导致(图4)。配合物的荧光相对于配体的荧光发生了明显的蓝移,其原因可能是由于形成配合物时金属离子与配体之间配位而影响配体分子内的电子跃迁,从而引起荧光蓝移[19]。
2.4 化合物的TG分析
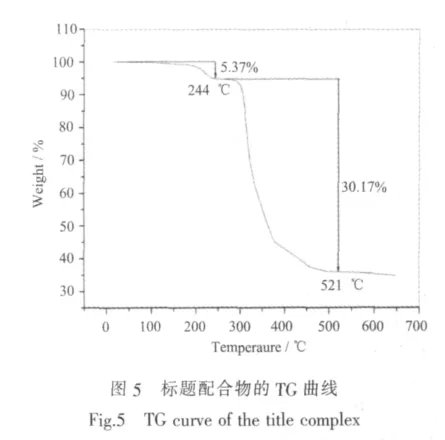
标题配合物的TG曲线如图5所示。从TG曲线可以看出,配合物从94.6℃开始失重,至244℃失重率为5.37%,相当于失去1个水分子(理论值为5.0%)。从275.6℃开始配合物分子骨架开始坍塌,至521℃出现平台,配合物失去有机配体L和草酸分子,最终残余物为CdO(实验值30.17%,理论值30.51%)。
2.5 化合物的XRD表征
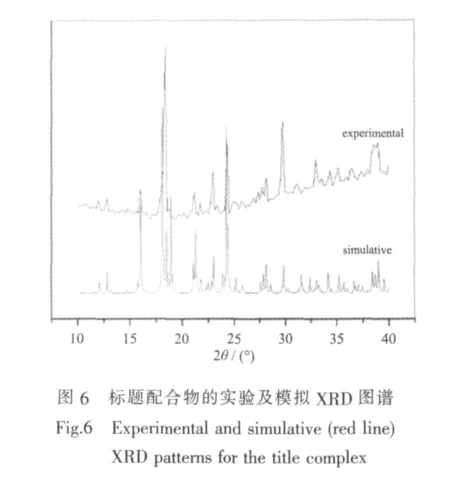
配合物的X-射线粉末衍射数据是在理学Ultima IV衍射仪上收集的,Cu Kα (λ=0.154 18 nm)射线,在 10°<2θ<40°范围内,如图 6 所示,实验测得的衍射图形与模拟的衍射曲线完全吻合,证实了化合物的相纯度。峰强度的不同可能是由于所测粉末样品的测定取向不同所致。
[1]Wang X L,Qin C,Wang E B,et al.Chem.Eur.J.,2006,12:2680-2691
[2]Pramanik S,Zheng C,Zhang X,et al.J.Am.Chem.Soc.,2011,133:4153-4155
[3]Lu Z Z,Zhang R,Li Y Z,et al.J.Am.Chem.Soc.,2011,133:4172-4174
[4]CHEN Lang(陈浪),CHENG Hui-Min(程慧敏),JIANG Jing-Jing(江静静),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,2:381-385
[5]Zheng B S,Bai J F,Duan J G,et al.J.Am.Chem.Soc.,2011,133:748-751
[6]ZHANG Zhong(张众),LIU Ji-Zhong(刘积中),GAO Peng(高鹏),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,1:195-200
[7]HAN Yin-Feng(韩银锋),LIU Tao(刘涛),DUAN Wen-Zeng(段 文 增 ),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28(1):149-152
[8]Miller J S.Adv.Mater.,2001,13:525-527
[9]Ma L Q,Lin W B.Angew.Chem.Int.Ed.,2009,48:3637-3640
[10]Batten S R.Crys Eng Comm,2001,3:67-72
[11]Friedrichs O D,O′Keeffe M,Yaghi O M.Solid State Sci.,2003,5:73-78
[12]HAN Yin-Feng(韩银锋),ZHENG Ze-Bao(郑泽宝),WU Ren-Tao(吴仁涛)et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(6):1125-1128
[13]ZHANG Peng(张鹏),WANG En-Bo(王恩波),XING Chang-Yu(邢长宇),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,3:560-562
[14]Zheng S T,Wu T,Chou C T,et al.J.Am.Chem.Soc.,2012,134:4517-4520
[15]Cao R G,Liu S X,Xie L H,et al.Inorg.Chem.,2007,46:3541-3547
[16]Fang R Q,Zhang X M.Inorg.Chem.,2006,45:4801-4810
[17]Fang R Q,Zhang X H,Zhang X M.Cryst.Growth Des.,2006,6:2637-2639
[18]LIU Hong-Wen(刘宏文),LU Wen-Guan(卢文贯).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27(11):2205-2210
[19]NIU He-Lin(牛和林),CHEN Ji-Tang(陈继堂),ZHANG Sheng-Yi(张胜义),et al.Chemical Research(Huaxue Yanjiu),2009,2:25-28
[20]Sheldrick G M.SHELXS-97 and SHELXL-97,Program for X-ray Crystal Structure Solution and Refinement,Gttingen University,Germany,1997.
[21]Zhang J Z,Cao W R,Chen Q W,et al.Inorg.Chem.Commun.,2007,10:1360-1364
