H2O-MgSO4C3H8体系中C3H8气体水合物分解动力学研究
2013-10-13李国恩陈盼盼曹吉林
李国恩,陈盼盼,赵 斌,曹吉林
(1.河北工业大学 河北省绿色化工与高效节能重点实验室,天津 300130;2.中国核电工程有限公司河北分公司,河北 石家庄,050011;3.天津市化工设计院,天津 300131)
0 前言
白钠镁矾(Na2SO4MgSO44H2O)资源在自然界储量巨大.从H2O-Na2SO4-MgSO4三元体系相图[1]分析可知,Na2SO4和MgSO4的分离比较适宜的方法是,先低温结晶提取Na2SO4,然后高温蒸发浓缩提取MgSO4,但是要想结晶析出MgSO4,必须浓缩除去溶液中大量水分,显然传统低温冷冻蒸发浓缩法是不经济的.气体水合物法分离技术是一种新型分离技术,具有水合物的生成条件温和、能耗低、分离效率高,对环境无害等优点.该技术已经在有机物分离[2]、溶液提浓[3]、气体分离[4]等领域进行了大量的研究,具有广阔的应用前景.基于对白钠镁矾资源的开发,作者提出了气体水合物法分离硫酸钠和硫酸镁的新思路,即白钠镁矾矿物经过水溶解除杂,低温结晶分离硫酸钠后,用气体水合物法浓缩富含硫酸镁母液,使硫酸镁析出制备硫酸镁,气体水合物分解后气体循环使用的工艺.
目前关于纯水体系中形成的C3H8气体水合物分解动力学研究,都是依据测定分解产生的气体量来进行数学关联和模型建立[5],气体测量受外界条件影响大,相对不及测定气体水合物分解的液相数据更加可靠.同时C3H8气体水合物浓缩分离含盐或非电解质水溶液时,形成的气体水合物都不同程度地存在母液夹带现象,采用测量气体法研究水合物的分解过程,难以了解水合物分解过程中母液夹带的变化规律,并且气体水合物在不同的溶液介质中形成固体结构也有差异.宋永臣等[6]发现阴离子、、Cl对甲烷合物分解条件的影响程度依次减弱,阳离子、、、的影响程度也依次减弱,且阴离子和阳离子对水合物分解的影响不同.Lu和Matsumoto[7]研究了不同组分孔隙水中水合物分解条件,发现阴离子和Cl明显影响其分解条件,而阳离子影响不明显.因此,研究三元体系中气体水合物分解过程,对气体水合物法分离白钠镁矾资源新工艺条件的制定具有重要指导作用.
1 实验
1.1 化学试剂
实验所用硫酸镁由天津市化学试剂一厂提供,纯度gt;99%;丙烷由天津伯克气体有限公司提供,纯度gt;99.9%;实验所用水均为二次去离子水,电导率lt;5 S cm1.
1.2 实验装置
本实验采用的相平衡测定装置参见图1,本实验采用恒定分解压力法.本实验采用的加压相平衡测定装置示意图见图1,主要包括透明高压反应釜、低温恒温装置、丙烷气瓶、加压泵、温度与压力测量仪表和数据采集系统等.实验系统的主要部件是带视窗的石英高压夹套反应釜,它所能承受的最大工作压力为1.6MPa,温度 15~100℃.透明反应釜内有一个三叶搅拌桨.釜的上部设有进气口、出气口、温度压力传感器接口和进料口,下部设有液体取样口、夹套冷却介质出入口、料液出口.反应器内的温度由其夹套内循环的低温冷却介质来控制,工作温度范围为 20~150℃,控温精度±0.01℃,用稀释的乙二醇溶液作为循环冷却介质,以保证反应所需的低温环境.实验温度由安装在反应釜内的Pt100铂电阻温度器测量,反应釜中的压力由0.25级精密压力表测定,量程为1.0MPa.
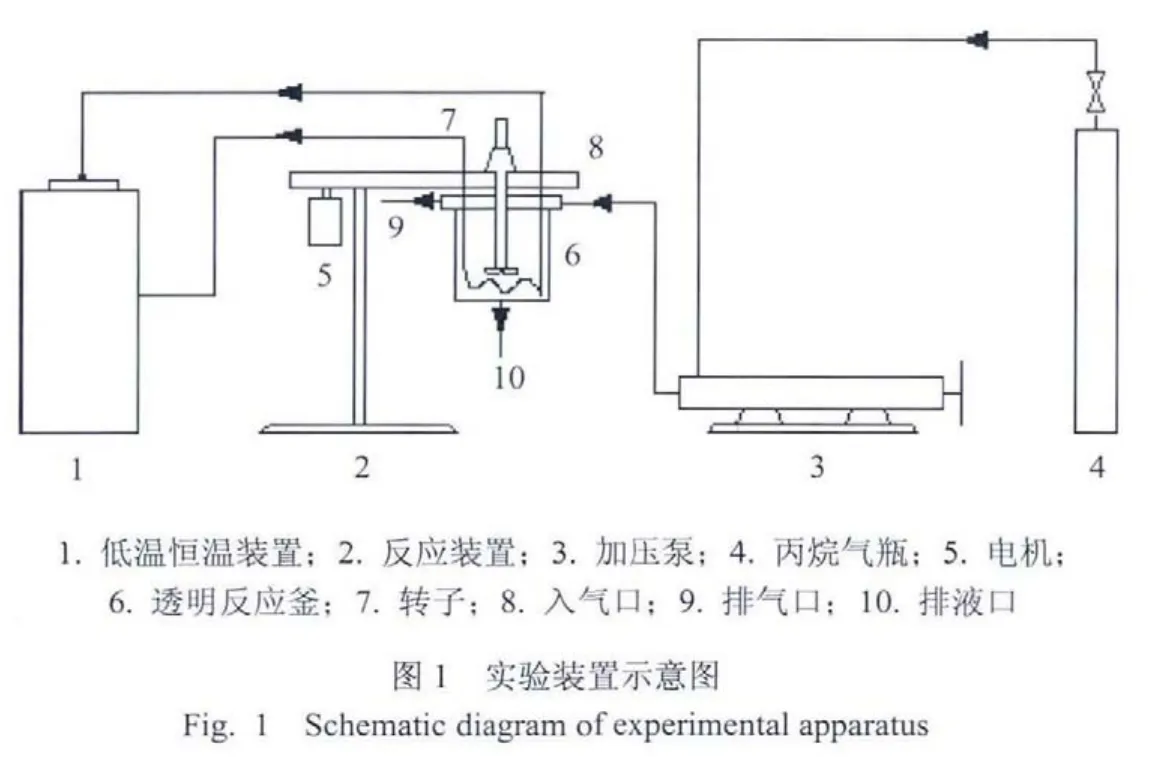
1.3 实验方法
配好一定浓度的MgSO4溶液,取出其中一部分准确称取质量后,注入干净的高压釜中,并开启搅拌.首先用C3H8气体置换釜内气体,待温度达到实验温度后,开始加压至0.4MPa左右.当透明釜中水合物已经生成,并且压力在1~2h不再变化,开始分解实验,先取出形成气体水合物后剩余的所有液相,然后将反应釜压力降至0.2MPa,并用加压泵维持反应釜内压力不变,开始计时,每隔20m in取出分解的液相,精确称量并分析其组成,直到反应釜中的固相完全溶解并全部取出后停止实验.以上过程维持温度为恒定值.
1.4 分析方法
Mg2+用EDTA络合滴定法,水的含量采用差减法.
2 实验结果
表1、表2、表3和表4分别为温度在276.15K、279.15K、281.15K和283.15K下H2O-MgSO4-C3H8三元体系中形成气体水合物分解过程的液固相组成变化实验数据.其中液相量及MgSO4含量是直接测得的,剩余的湿固相量及其组成是由开始加入物料量减去取出的液相量计算得出,湿固相夹带母液量依据 MgSO4不形成水合物、湿固相中所含MgSO4全部来源于母液的原理,按“母液夹带量=湿固相总量×湿固相中MgSO4含量/取出液相中MgSO4含量”计算得出,以水计的水合物量由湿固相总量减去夹带母液量得出,湿固相中形成水合物量(包含 C3H8)是以水计的水合物量为基准,按 C3H8气体分子与水分子摩尔比计算得到[8-9];值是以水计的水合物与夹带母液质量之比.从分解温度和压力达到实验条件时开始计时.0 m in时剩余固相量即为分解实验开始时的初始固相量.
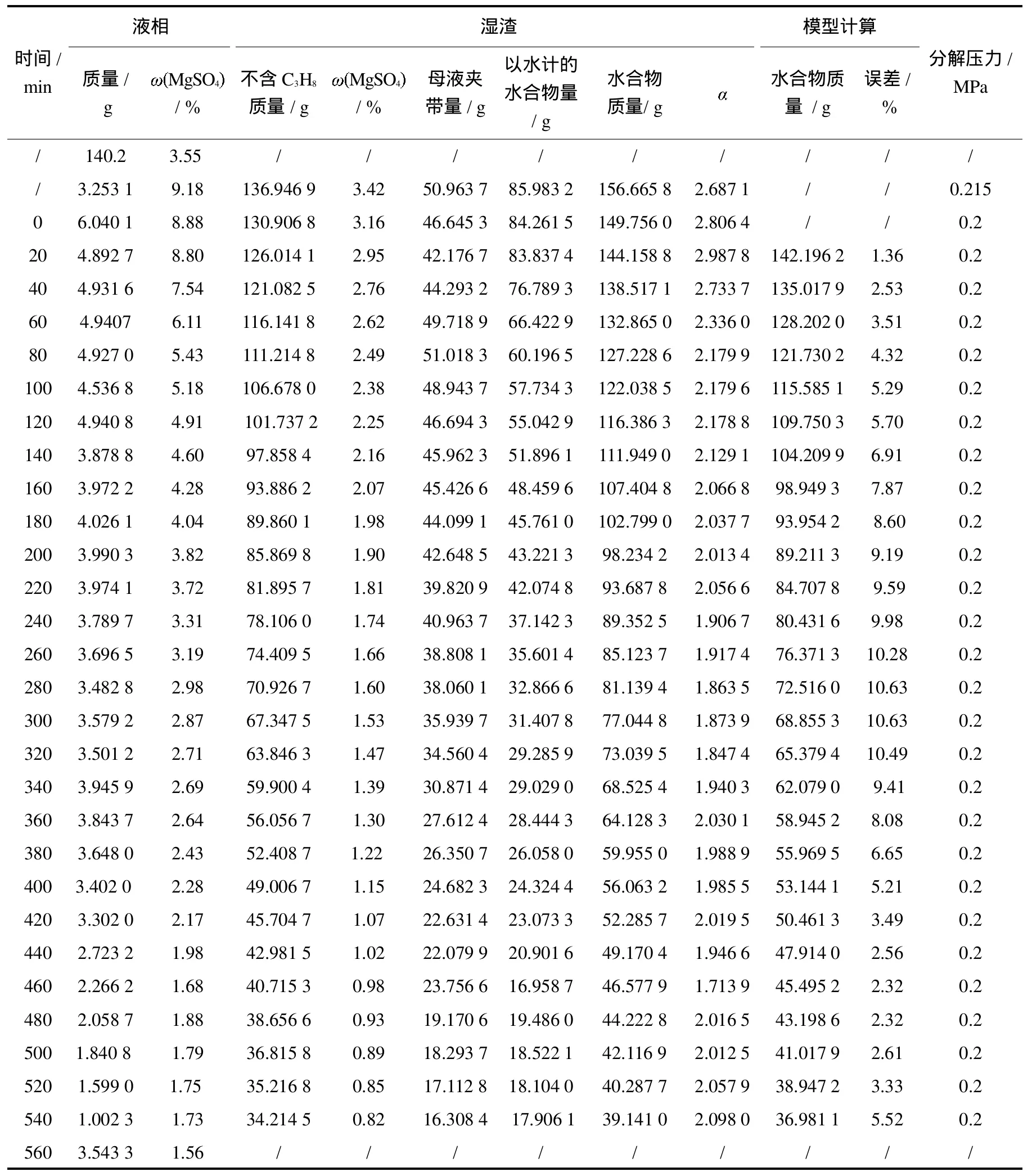
表1 276.15 K分解实验数据Tab.1 The decomposition data at276.15 K
由表1、表2、表3和表4可以看出,不同初始原料组成下形成的水合物量不同.当MgSO4浓度增加时,形成水合物的平衡压力也升高.从水合物分解过程中剩余湿固相的 值变化规律可以看出,随着水合物的分解,剩余水合物量越来越少,母液夹带量则逐渐增高,说明水合物分解时结构变得越来越疏松,母液夹带位于水合物晶粒之间.图2为不同温度下,C3H8气体水合物分解过程中液相硫酸镁浓度与水合物中硫酸镁浓度的变化关系.由图2可以看出随着水合物的分解,剩余固相中液相中MgSO4含量和分解的液相中MgSO4含量比值在降低,硫酸镁的夹带量变大,为工艺制定提供基础数据.
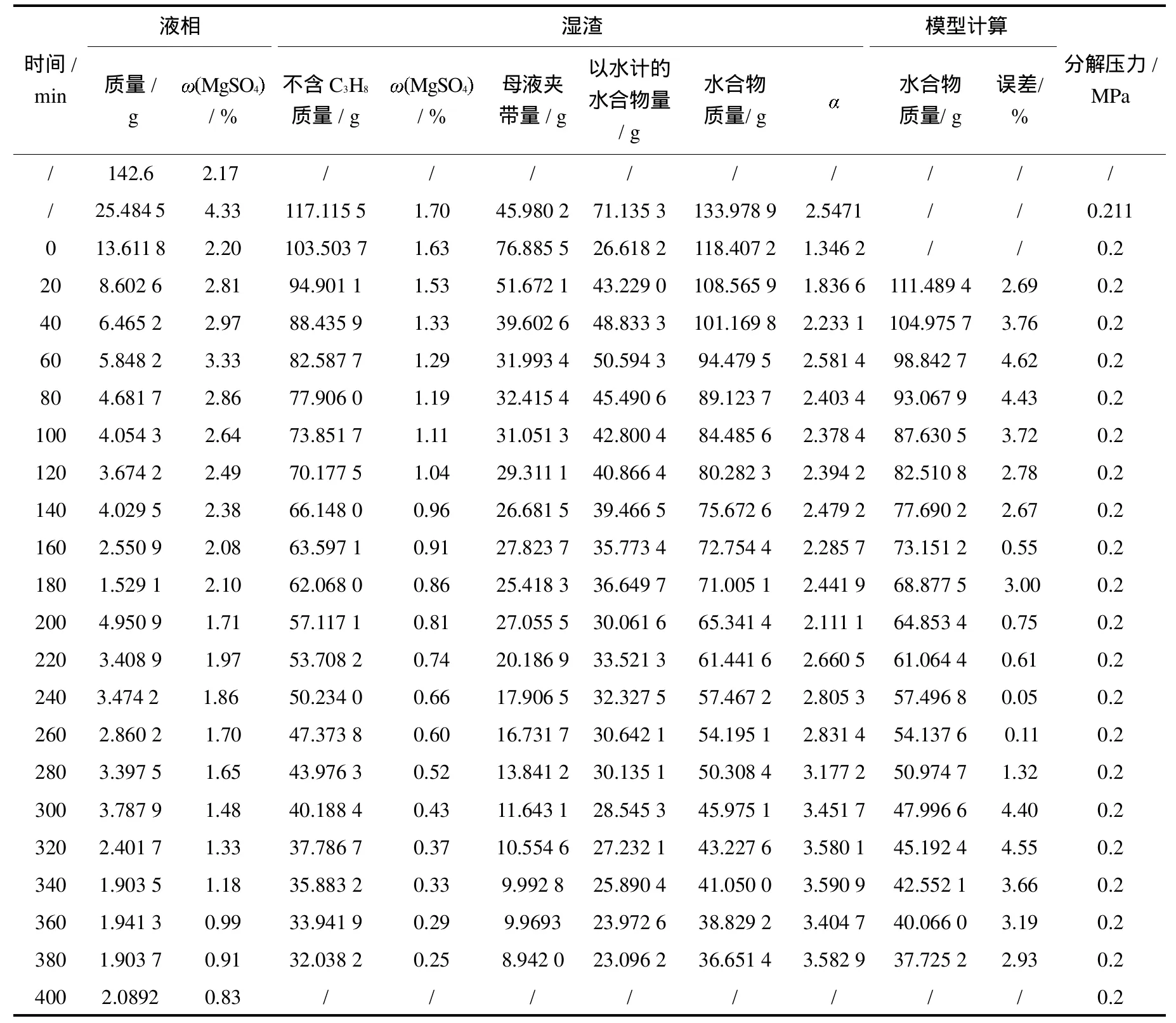
表2 279.15 K分解实验数据Tab.2 The decomposition data at279.15K

表3 281.15 K分解实验数据Tab.3 The decomposition data at281.15 K
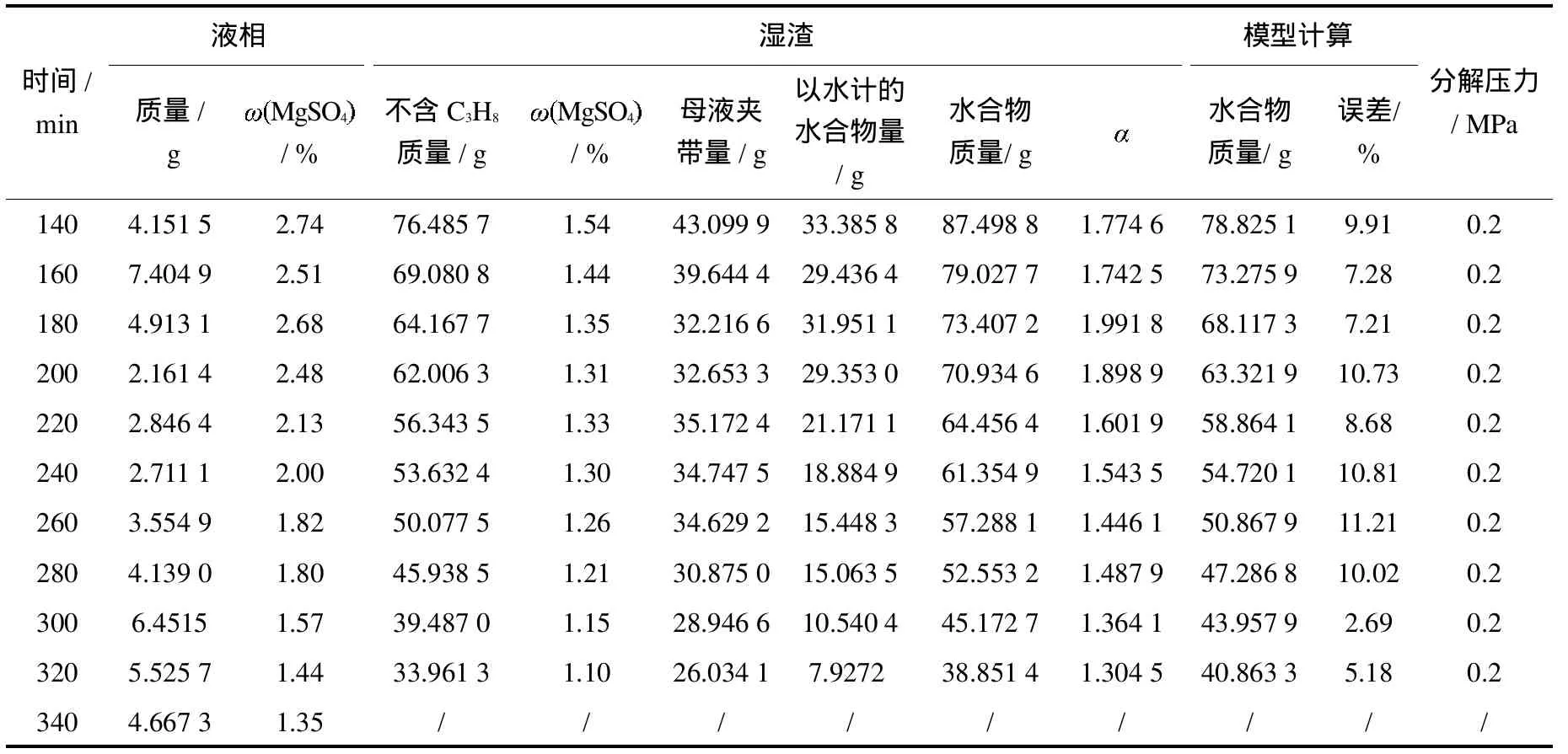
续表3 281.15 K分解实验数据Tab.3 The decomposition data at281.15 K(continued)

表4 283.15 K分解实验数据Tab.4 The decomposition data at283.15 K
3 分解动力学分析
3.1 分解动力学模型及水合物分解速率常数
本实验体系中水形成气体水合物的分解反应可用下式表述


图2 C3H8气体水合物分解过程中液相和水合物中硫酸镁浓度的变化关系Fig.2 The relationship between the concentration ofmagnesium sulfate in the liquid phaseand thehydrate phase during the decomposition of C3H8 gashydrate
其中: 代表笼型化合物中每个水分子所包络的气体分子数.温度、压力、气体水合物粒子表面积和分解推动力等因素对气体水合物分解速率都有很大影响.气体水合物分解过程可以分为气体水合物结构的解构和C3H8分子从固体表面解吸2个步骤,即为水合物粒子表面笼型主体晶格破裂,粒子收缩,客体分子从表面逸出[10].本实验分解温度都在0℃以上,当气体水合物分解产生的C3H8进入高压釜上部气相后,分解产生的水也变成液态与水合物固相分离,进入夹带的母液中并流向高压釜底部,因此气体水合物分解过程可以不考虑质量传递的影响.气体水合物分解是在恒压恒温下进行,C3H8气体逸度C3H8即为定值,此值可采用已给定的温度和平衡压力值SRK方程计算得出[11].给定温度下与气体水合物平衡的逸度 也为定值,此值也可采用SRK方程计算得出.所以气体水合物分解推动力 = 也为定值.假定气体水合物由均匀的粒子构成,则给定温度压力条件下气体水合物的分解速率r与水合物质量w成正比,r可写作

当t=0时,w=w0,式中w0和k'分别为分解实验开始时形成气体水合物的质量与表观分解速率常数.对式(1)积分可得

表观分解速率常数k'与分解推动力 有关,可用如下线性关系表示

其中 为本征分解速率常数,与推动力大小无关.根据实验数据拟合的k值可预测C3H8气体水合物在其他温度和推动力下的表观分解速率常数.依据方程式 (2)和 (3)对表1~4中实验数据进行拟合,可以求得k'进而可进行分解过程计算,计算结果见表1~4,模型计算值与实验值吻合良好,相对误差大部分在10%以内.误差产生的原因一是因为该模型忽略了气相主体到粒子表面的传质阻力和水相主体到粒子表面的传热阻力;二是因为随着实验的进行,气体水合物固相逐渐变小,尤其到最后阶段和釜壁接触不是很充分从而引起误差.
表5为各分解温度下分解速率常数.由表可知,体系的平衡压力受进料 MgSO4浓度影响较大,MgSO4浓度越高,平衡压力越大.水合物分解温度越高,则水合物分解越快;推动力越大,则水合物分解越快.水合物分解速率由实验分解温度和分解压力共同决定,分解温度起主导作用.
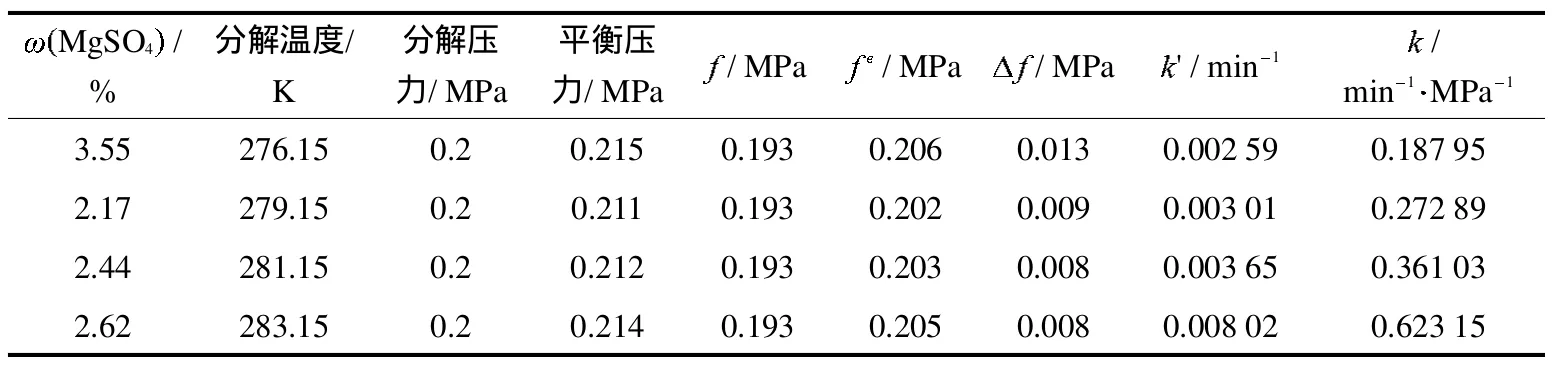
表5 分解速率常数Tab.5 The decomposition rate constants
3.2 气体水合物分解活化能 Ea的确定
理想情况下,式 (3)中速率常数k可采用Arrhenius方程[12]描述
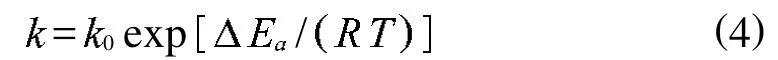
其中:0为笼型化合物本征分解速率常数,m in1MPa1,类似于Arrhenius方程中的指前因子,与压力、温度无关,取决于初始粒子的几何形状; Ea为活化能,kJ/mol;T为温度,K.由表5中数据以lnk对1作图见图3,得到一条直线,经线性回归后计算可得 Ea=107.489 kJ/mol.
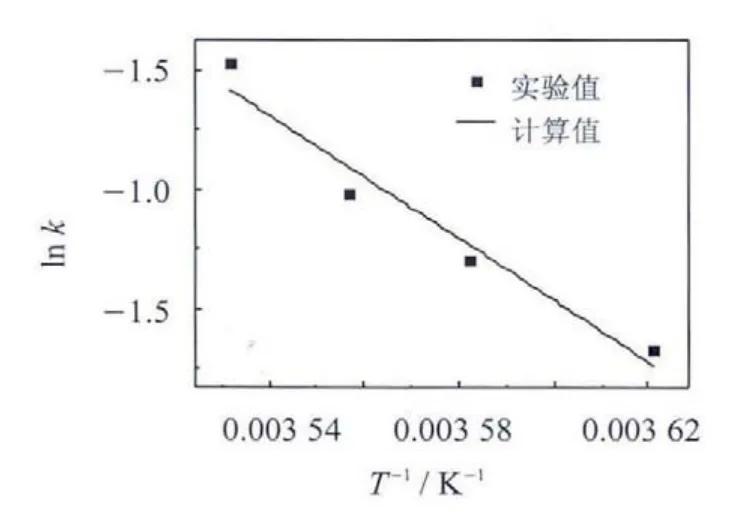
图3 分解活化能的拟合曲线Fig.3 The fitted curveof activation energ
4 结论
非纯水体系H2O-MgSO4-C3H8中形成的C3H8气体水合物分解过程的实验数据表明,恒温恒压条件下,随气体水合物分解,母液夹带量增多,水合物变得越来越疏松.依据实验数据建立的 H2O-MgSO4-C3H8三元体系中形成C3H8气体水合物的分解动力学模型,其计算值与实验值吻合良好;276.15 K,279.15 K,281.15 K和283.15 K条件下C3H8气体水合物的表观分解速率常数和本征分解速率常数,随温度升高逐渐增加.C3H8气体水合物的分解活化能 Ea=107.489 kJ/mol.
[1]余杰,陈美珍.Na2SO4-MgSO4-H2O体系的相图与两盐分离方法的研究 [J].海湖盐与化工,1994(4):32-36.
[2]Yoon Ji-Ho,Lee H.Clathrate phasee quilibria for the water-phenol-carbon dioxide system[J].AIChE J,1997,43(7):1884.
[3]Willson RC,BulotE,Cooney C L.Useof hydrates for aqueous solution treatment:US,4678583[P].1987-07-07.
[4]DouglasG,Elliot Houston.Process for separating selected components frommulti-componentnatural gas streams:US,5660603[P].1997-08-26.
[5]Jamaluddin A KM,KalogerakisN,BishnoiPR.Modeling of decomposition of asynthetic core of me than egashy drateby coupling intrinsic kinetics w ith heat transfer rates[J].PhysChem,1989,67(6):948-954.
[6]宋永臣,杨明军,刘瑜,等.离子对甲烷水合物相平衡的影响 [J].化工学报,2009,60(6):1362-1366.
[7]孙始财,刘昌岭,业渝光,等.氯盐溶液中甲烷水合物高压分解条件及影响因素 [J].物理化学学报,2011,27(12):2773-2778.
[8]刘海彬,曹吉林,郭康宁.高压低温下H2O-H2O2-CO2三元体系CO2气体水合物的分解动力学 [J].过程工程学报,2010,10(2):282-286.
[9]Gnanendran N,Amin R.Equilibrium hydrate formation conditions for hydrotrope-water-natural gassystems[J].Fluid Phase Equilibria,2004,221(2):175-187.
[10]Kim H C,BishnoiPR,Heidemann R A,etal.Kinetics of methanehy dratede composition[J].Chem Eng Sci,1987,42(7):1645-1653.
[11]朱自强,徐汛.化工热力学 [M].北京:化学工业出版社,1991:152-155.
[12]ClarkeM,BishnoiPR.Determ inationof theintrinsic rate of e thane gashy dratedecomposition[J].Chem Eng Sci,2000,55(21):4869-4883.
