均苯四甲酰亚胺桥联的聚酞菁亚铁的氧还原反应
2013-09-21孙晓然李光跃夏定国张立美
孙晓然 李光跃 夏定国 张立美 李 钒
(1北京工业大学化学与环境工程学院,北京100086;2北京大学工学院,北京100871;3河北联合大学化学工程学院,河北唐山063009)
1 Introduction
In the past decades,phthalocyanine(Pc)has been widely applied in many fields,1including dyeing,2,3chemical sensors for gases,4liquid crystals,5,6nonlinear optics,7and catalysis.8Specificity in the applications of Pcs can be introduced by modification of the Pc ring or changes in the central metal.Pcs can form coordination complexes with most metal elements.These complexes are also intensely colored and are thus used as dyes or pigments.2,3Due to their large conjugated systems,they exhibit distinct optical and electrical properties,good chemical stability,and excellent electrocatalysis activities.In recent years,Pcs as the cathode catalysts have gradually substituted for the Pt/C electrode in direct-methanol fuel cells(DMFCs).
DMFCs are a kind of proton-exchange fuel cells in which methanol is used as the fuel.Their main advantage is easy transportation of methanol,which are energy-dense and stable at all environmental conditions.At present,the efficiency of these cells is quite low.So they are targeted especially to portable applications,where energy and power density are more important than efficiency.Yet,methanol crossover is a problem for the development of DMFCs.This problem can be solved by two ways:(1)preparing the proton-exchange membrane with low methanol-transmittance;(2)preparing new electrocatalysts resistant in methanol oxidation.Nowadays,the methanol-transmittance of the proton-exchange membrane is still very high,which could significantly reduce the performance of DMFCs.Polymeric metallophthalocyanines are electrocatalytically active in oxygen-reduction and resistant in methanol-oxidation.As the cathode catalysts of DMFCs,they draw more and more interests in the research.

Fig.1 Structure of poly-PcFe
Fundamentally,redox reactions are a family of reactions that are concerned with the electron or charge transfer between molecules.9In order to better understand the redox mechanism,there have been a lot of efforts to complement experiments by theory.Recently,charge transfer and some other electronic processes have started to be studied computationally for conjugated molecules and their related systems.10-17Based on the ideas mentioned above,we designed a new kind of pyromellitimide-bridged polyphthalocyanine Fe(II)(poly-PcFe,shown in Fig.1)catalyst.This catalyst can be easily synthesized.It is important to understand the reaction process of O2reduction because of its crucial role in determining electrocatalysis activity.In order to provide a complementary view on the O2-reduction reaction,we performed a density functional theory(DFT)to study concerning this catalyst.Three model molecules are designed with different polymerization degrees(P1,P2,and P3,which are shown in Fig.2).To evaluate the influence of polymerization degrees on the O2reduction,molecular geometries were optimized for P1,P2,and P3.In particular,we focused our attention on the frontier molecular orbitals in the model molecules and O2,and the polymerization degree effect on the O2reduction.By scanning the potential curves of the possible process in the O2reduction,we can obtain the trend of reactions between O2and poly-PcFe,thus explaining the process of the O2-reduction reaction.
2 Computational methods
All calculations on electronic structures were carried out using the ORCA 2.8 program.18Geometry optimizations and vibration analysis were performed using density functional theory(DFT)method.In DFT,the electron density played the key role and all molecular properties could be derived from the density.The computational effort for DFT was much smaller than for correlated ab initio treatments and actually quite similar to Hatree-Fock theory.DFT applications required the choice of a suitable exchange-correlation functional and one-electron basis set.In this work,a test with a series of functionals(BP86,BLYP,PW91,B3P86,B3LYP,and B3PW91)was performed to confirm the proper functional.As the result,BP86 functional19,20was chosen as the functional,which was also considered to be the preferred DFT approach for carrying out related research.21The single valence quality with one set of polarization functions(SVP)22was chosen as basis sets throughout,which was a proper basis set for ionic compound23,24and not time-consuming.No constrains for symmetry,bonds,angles,or dihedral angles were applied in the geometry optimization calculations.All of the local minima were confirmed by the absence of an imaginary mode in vibrational analysis calculations.Furthermore,the natural bond orbital(NBO)analysis was performed using the NBO 3.0 program.25A useful aspect of the NBO method was that it gives information about interaction in both filled and virtual orbital spaces.26Moreover,the potential energy curves were qualitatively scanned by constrained optimizations,keeping the target bond distance fixed at a serious of values.According to our previous report,27the energy of reaction P+H→PH could take place of the energy of reactionPH in the calculations.
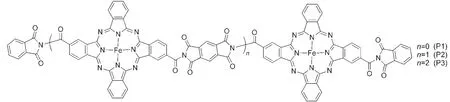
Fig.2 Structures of three models(P1,P2,and P3)of poly-PcFe
3 Results and discussion
3.1 Molecular geometries
The optimized geometries of pyromellitimide-bridged polyphthalocyanine Fe(II)with different polymerization degrees(n=1-3)are obtained using the BP86 functional with SVP basis sets(shown in Fig.3,P1,P2,and P3 were described in this paper).Their spin multiplicities(S)are 5,9,and 13,respectively.Because of large conjugation effect,all the atoms in phthalocyanine moieties of P1,P2,and P3 are coplanar.Four Fe―N bonds are all 0.1985 nm in P1,which indicates that the Fe―N bonds are equivalent and the dicarboximino groups have little effect on the molecular structure of P1.But pyromellitimide moieties do not conjugate with a benzene ring in the three molecules,due to the stereo-hindrance effect.They have a dihedral angle of 75°.All the bond lengths of Fe―N are 0.1981-0.1982 nm in P2 and P3,slightly shortened by the pyromellitimide moieties.
We also optimized the geometries of the complexes formed by the three poly-PcFe models and O2.The results show that their geometries are all similar.The complex P3-O2is shown in Fig.4.In this molecule,the bond angle of Fe―O―O is 121°.The Fe―O bond length is 0.228 nm,shorter than the sum of the van der Waals radii(0.278 nm)for the corresponding atoms.28So,the Fe atom in P3 can interact with O2with a weak interaction.According to the Mulliken charge analysis,both of the O atoms are negatively charged.The O―O bond length is 0.126 nm,longer than that in the O2molecule(0.121 nm).This indicates the donation of electrons from the central Fe atom to the anti-bonding orbital of O2.According to the NBO charge analysis,the natural charges of two O atoms in P3-O2are-0.106e and-0.092e,respectively.Thus,P3-O2can be considered as P3 binding an O2molecule.
3.2 Stability of the O2-complexes

Fig.4 Geometry of P3-O2complex
We also calculated the binding energy(ΔE)for the reaction between O2and the three poly-PcFe models to determine the stability of their O2-complexes.As shown in Fig.5,it can be seen that each reaction has a negative binding energy,indicating that all of the calculated reactions are exothermic.That is to say,the selected O2-complex structures are all stable.It is evident that the ΔE decreases almost linearly with increasing polymerization degree.Accordingly,poly-PcFe models with larger polymerization degree should have better stability.
3.3 Molecular orbital analysis
According to the study on metallophthalocyanines of Zagal et al.,29,30the interaction between molecular orbital of O2and the atomic orbital of the central metal could cause the charge transfer from the metal atom to O2.The O2-reduction process in this work can be shown as:

Frontier molecular orbital(FMO)theory has recently provided an elegant explanation for the redox reactions.31In this theory,the lowest unoccupied molecular orbital(LUMO),the highest occupied molecular orbital(HOMO),and the singly occupied molecular orbital(SOMO)play vital roles.For the O2-reduction reaction of poly-PcFe,the following criteria must be met:(1)the molecular orbitals must align for good overlap,which can explain why the Fe―O―O bond angle is nearly 120°;(2)the energies of the SOMOs in O2must be lower than those of the HOMOs in the poly-PcFe models,which is necessary for O2-reduction.As shown in Fig.6,the energy of HOMO in the poly-PcFe models,is higher than that of the SOMOs of O2.This demonstrates that all of the poly-PcFe models can be applied to the catalysis of O2-reduction.It is worthwhile to note that the HOMO energy is increased as the polymerization degree of poly-PcFe models is increased.Comparing the FMO energies,we can conclude that the substituent groups in phthalocyanine Fe(II)polymers could influence their electrocatalytic activities.Stronger electron-withdrawing groups could increase the electrocatalytic activity of Fe-phthalocyanine polymers,such as pyromellitimide groups.This agrees with the studies by Zajal et al.30
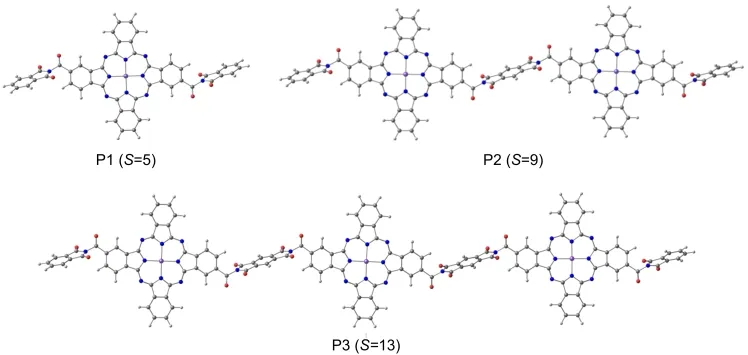
Fig.3 Geometries of P1,P2,and P3

Fig.5 Calculated binding energies of the O2-complexes of poly-PcFe models
As reported,Eckhardt et al.32described the relationship between the frontier-orbital energy of conjugated polymers and the onset redox potential for the first time.The formula proposed by them can be written as follows:
EHOMO=-e(φonset,re+4.4)(unit:eV)
where EHOMO,e,and φonset,restand for the energy of HOMO,electron charge,and onset reduction potential,respectively.According to the HOMO energy of the poly-PcFe models,we can estimate that the onset potential of O2reduction is higher than 0.87 eV.Furthermore,the results show that poly-PcFe with higher polymerization degree exhibits stronger reduction ability.
Molecular orbital diagram is an appropriate tool in explaining chemical bonding in molecules.33The molecular orbital diagrams have been calculated for P1-O2.As shown in Fig.7,a σ-bonding orbital can be formed by the linear combination of dz2orbital of Fe atom and π*2pyorbital of O2molecule;a π-bonding orbital can be formed by the linear combination of dyzorbital of Fe atom and π*2pyorbital of O2molecule.In other words,the Fe atom connects with O2molecule by a double bond.Meanwhile,the NBO analysis also confirms the double bond.But the occupancies of them are 0.324 and 0.238.This confirms the weak interaction between Fe atom and O2molecule.
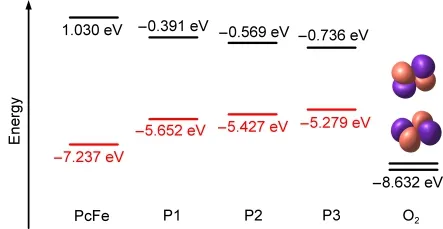
Fig.6 Calculated energy levels of HOMOs,LUMOs,and SOMOs
3.4 Potential energy curves and O2-reduction electrocatalytic cycle
The potential energy curves of P1 involved in the process of the O2-reduction reaction have been studied.Although the DFT method cannot be expected to be sufficiently accurate in calculating energies,previous calculations have indicated that the method may be reliable in calculating hydrogen-transfer potential energy.15,17,18Fig.8(A)shows that the combination of the Fe atom in P1 and O2molecule is an energetically favorable process with a very low potential barrier.The displacement of water molecule by O2could be the rate-determining step in the catalytic cycle.34Fig.8(B)shows a barrierless potential-energy curve for P1-OOH model,the energy of which decreases with the O―H bond length shortened from 0.287 to 0.097 nm.This indicates that the complex P1-O2can spontaneously capture the H+cation in the acidic solution and be reduced to P1-OOH.Meanwhile,Fe(II)is oxidized to Fe(III).As depicted in Fig.8(C),P1-OOH changes into P1-OOH2by capturing the H+cation,which is also a spontaneous exothermic process.Its binding energy is smaller than that of P1-O2,because capturing the first H+cation could destroy the O=O double bond in P1-O2and thus need more energy.The data in Fig.8(D)confirmed that P1-OOH2is unstable.It could decompose into P1-O and a water molecule when Fe(III)is oxidized to Fe(IV).Actually,the H+-capturing of P1-OOH and the water-disintegration from P1-OOH2are simultaneous.Figs.8(E)and 8(F)provide the potential energy curves of H+-capturing process of P1-O as functions of the corresponding O―H bond lengths.Similar with P1-O2,P1-O can spontaneously be reduced to P1-OH2,which could be considered as P1 binding a water molecule.Fe(IV)is gradually reduced to Fe(II)in the process.

Fig.7 Molecular orbitals formed by the atomic orbitals in Fe and O2in P1
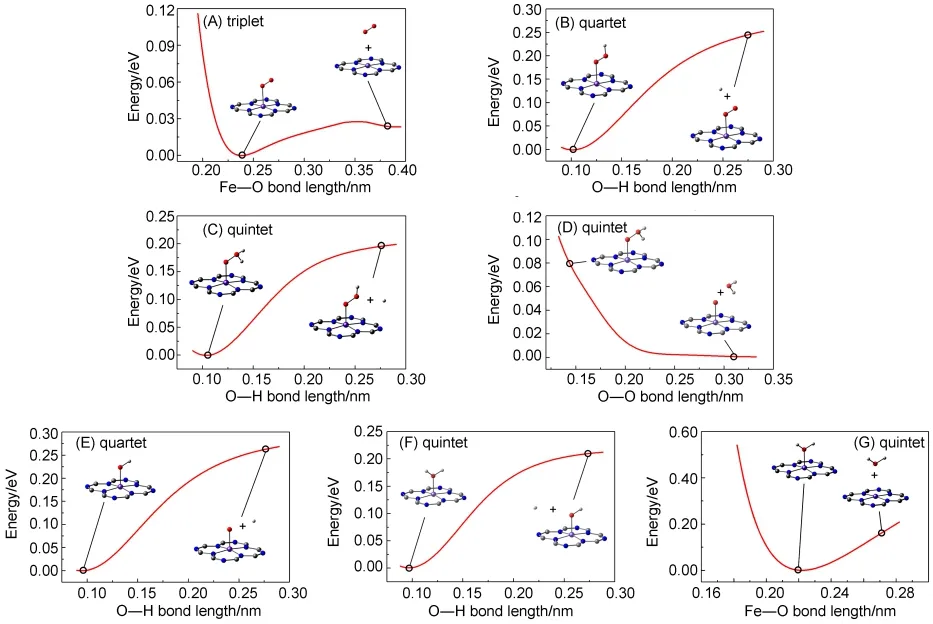
Fig.8 Calculated potential energy curves involved in the O2-reduction reaction
Fig.9 depicts the O2-reduction reaction of poly-PcFe in details,where the ring of the Pc is represented by two bold vertical lines.Poly-PcFe is used in O2-reduction electrodes that operate by means of the electrocatalytic cycle.The cycle begins with the molecule P1,in which Fe(II)atom is tetra-coordinated by Pc ring.Subsequent binding of O2yields the Fe(II)-O2complex(triplet),a good electron acceptor.This could trigger the reduction of O2(step 1).Since the Fe(II)-O2is a good Lewis base,it captures a proton to form the Fe(III)-hydroperoxide species(quartet,step 2).The Fe(III)-hydroperoxide species is still a good Lewis base and captures another proton.When the cathode supplies a electron,a Fe(IV)-O complex(quintet)and a water are formed(steps 3 and 4).Then,the oxygen atom in the Fe(IV)-O complex is protonated.In this case,the oxygen atom is converted into another water molecule.In the meantime,the Fe(IV)is reduced to Fe(II)(steps 5 and 6,Fe(III)-OH:quartet;Fe(II)-OH2:quintet).After this electrocatalytic reaction,the O2molecule can be reduced to two water molecules in the acidic solution,and the poly-PcFe restores to its beginning state by releasing a water molecule.
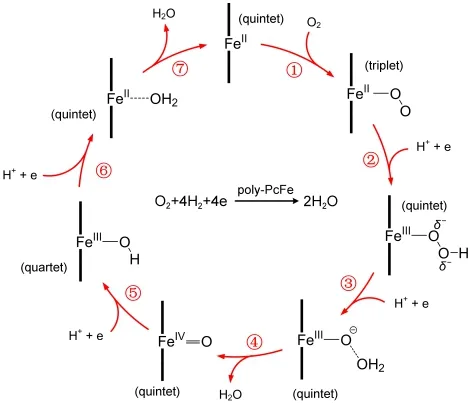
Fig.9 Schematic representation of the electrocatalytic cycle of poly-PcFe
4 Conclusions
In summary,the process for O2reduction of poly-PcFe was studied by DFT method.Structual analysis and FMO analysis by DFT calculations confirm that the pyromellitimide moieties and phthalocyanine moieties of poly-PcFe are not coplanar.The Fe atom in pyromellitimide moiety could combine O2and form a stable complex by a double bond.The double bond is a linear combination of the d orbital of Fe and SOMO of O2.The charge transfer process takes place from the Fe atom to the binding O2molecule.The FMO results also show that the HOMO energy is increased as the polymerization degree of poly-PcFe isincreased,and strong electron-withdrawing groups of poly-PcFe are favorable to O2reduction.So,the poly-PcFe catalyst with higher polymerization degree and stronger electron-withdrawing groups could have better activi-ties of O2reduction.Based on the HOMO energies,the onset potential of O2reduction with the catalyst is estimated to be less than 0.87 eV.The potential energy curves confirme that O2can be reduced on the poly-PcFe catalyst by means of an electrocatalytic cycle.This poly-PcFe catalyst has electrocatalytic activity for O2reduction in acidic medium.
(1) Phthalocyanines-Properties and Applications;Leznoff,C.C.,Lever,A.B.P.Eds.;VCH:New York,1989-1996.
(2) McKeown,N.B.Phthalocyanine Materials-Synthesis,Structure and Function;Cambridge University Press:Cambridge,1998.
(3)The Porphyrin Handbook;Kadish,K.,Smith,K.M.,Guilard,R.Eds.;Academic Press:San Diego,2003;Vols.15-20.
(4) de la Torre,G.;Vasquez,P.;Agulló-López,F.Adv.Mater.1997,9,265.
(5) Simon,J.;Sirlin,C.Pure Appl.Chem.1989,61,1625.doi:10.1351/pac198961091625
(6) Humberstone,P.;Clarkson,G.J.;McKeown,N.B.;Treacher,K.E.J.Mater.Chem.1996,6,315.doi:10.1039/jm9960600315
(7) Dini,D.;Hanack,M.Physical Properties of 107 Phthalocyanine-Based Materials.In The Porphyrin Handbook,Volumes 11-20:Phthalocyanines:Properties and Materials;Academic Press:San Diego,2003;Vol.17,p 1.
(8) Lever,A.B.P.;Hempstead,M.R.;Leznoff,C.C.;Liu,W.;Melnik,M.;Nevin,W.A.;Seymour,P.Pure Appl.Chem.1986,58,1467.doi:10.1351/pac198658111467
(9) Sirotin,S.V.;Tolbin,A.Y.;Moskovskaya,I.F.;Abramchuk,S.S.;Tomilova,L.G.;Romanovsky,B.V.J.Mol.Cat.A:Chem.2010,319,39.doi:10.1016/j.molcata.2009.11.017
(10) Han,K.L.;He,G.Z.J.Photochem.Photobiol.C 2007,8,55.doi:10.1016/j.jphotochemrev.2007.03.002
(11) Zhao,G.J.;Han,K.L.J.Phys.Chem.A 2007,111,2469.doi:10.1021/jp068420j
(12) Zhao,G.J.;Han,K.L.Biophys.J.2008,94,38.doi:10.1529/biophysj.107.113738
(13)Yu,F.B.;Li,P.;Li,G.Y.;Zhao,G.J.;Chu,T.S.;Han,K.L.J.Am.Chem.Soc.2011,133,11030.doi:10.1021/ja202582x
(14) Li,G.Y.;Zhao,G.J.;Liu,Y.H.;Han,K.L.;He,G.Z.J.Comput.Chem.2010,31,1759.
(15) Zhao,G.J.;Han,K.L.Accounts Chem.Res.2012,45,404.doi:10.1021/ar200135h
(16) Li,G.Y.;Chu,T.S.Phys.Chem.Chem.Phys.2011,13,20766.doi:10.1039/c1cp21470e
(17)Li,G.Y.;Zhao,G.J.;Han,K.L.;He,G.Z.J.Comput.Chem.2011,32,668.doi:10.1002/jcc.v32.4
(18) Neese,F.ORCA-an Ab initio,Density Functional and Semiempirical Program Package;2008.http://www.thch.uni-bonn.de/tc/orca/
(19) Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/1.464913
(20) Perdew,J.P.Phys.Rev.B 1986,33,8822.doi:10.1103/PhysRevB.33.8822
(21) Himo,F.;Siegbahn,P.E.M.Chem.Rev.2003,103,2421.doi:10.1021/cr020436s
(22)Treutler,O.;Ahlrichs,R.J.Chem.Phys.1995,102,346.
(23) Zhong,A.G.;Huang,L.;Jiang,H.J.Acta Phys.-Chim.Sin.2011,27,837.[钟爱国,黄 凌,蒋华江.物理化学学报,2011,27,837.]doi:10.3866/PKU.WHXB20110323
(24) Schneider,S.K.;Julius,G.R.;Loschen,C.;Raubenheimer,H.G.;Frenking,G.;Herrmann,W.A.Dalton Trans.2006,1226.
(25)http://www.ccl.net/cca/software/SOURCES/FORTRAN/nbo/index.shtml
(26) Zheng,W.R.;Xu,J.L.;Xiong,R.Acta Phys.-Chim.Sin.2010,26,2535.[郑文锐,徐菁利,熊 瑞.物理化学学报,2010,26,2535.]doi:10.3866/PKU.WHXB20100931
(27) Chen,X.;Li,F.;Wang,X.Y.;Sun,S.R.;Xia,D.G.J.Phys.Chem.C 2012,116,12553.doi:10.1021/jp300638e
(28) Favia,A.D.;Cavalli,A.;Masetti,M.;Carotti,A.;Recanatini,M.Proteins 2006,62,1074.
(29) Zagal,J.H.;Cárdenas-Jirón,G.I.J.Electroanal.Chem.2000,489,96.doi:10.1016/S0022-0728(00)00209-6
(30) Zagal,J.H.;Gulppi,M.;Issacs,M.;Cardenas-Jiron,G.;Aguirre,M.J.Electrochim.Acta 1998,44,1349.doi:10.1016/S0013-4686(98)00257-6
(31) Fendorf,S.E.;Fendorf,M.;Sparks,D.L.;Gronsky,R.J.Colloid Interface Sci.1992,153,37.doi:10.1016/0021-9797(92)90296-X
(32) Eckhardt,H.;Shacklette,L.W.;Jen,K.Y.J.Chem.Phys.1989,91,1303.doi:10.1063/1.457153
(33) Jean,Y.;Volatron,F.An Introduction to Molecular Orbitals;Oxford University Press:Oxford,1993.
(34) Shaik,S.;Kumar,D.;de Visser,S.P.;Altun,A.;Thiel,W.Chem.Rev.2005,105,2279.doi:10.1021/cr030722j
