芳基磺酰胺类丙酮酸激酶M2激动剂的基于多复合物的药效团模型和定量构效关系
2013-09-17陈正军蒋庆琳
陈正军 蒋庆琳 何 谷 韩 波 郭 丽
(1四川大学华西药学院,成都610041;2成都医学院药学系,成都610500;3四川大学华西医院,生物治疗国家重点实验室,成都610041;4成都中医药大学药学院,成都611137)
1 引言
肿瘤细胞的能量主要来自糖酵解,肿瘤细胞的细胞能量代谢特性对于肿瘤的进展起着关键作用,1-4德国科学家Otto Warburg早在上世纪20年代就提出了著名的瓦伯格效应:肿瘤细胞比正常细胞需要更多的能量和核酸维持其生长增殖,而能量代谢不但为肿瘤细胞的生长提供能量,而且为肿瘤细胞增殖所需核酸的合成提供原材料,即使在有氧情况下,肿瘤细胞仍偏好于糖酵解方式进行葡萄糖代谢,而不采用能产生更多ATP的线粒体氧化磷酸化方式进行能量代谢.5,6随着近年来氟化去氧葡萄糖正电子摄影断层扫描(FDG-PET)技术的广泛应用,组织标本的葡萄糖摄取量可检测并成像,瓦伯格效应在越来越多的肿瘤类型中得以证实,探索通过阻断糖酵解途径而抑制癌细胞中能量的生成从而治疗恶性肿瘤的策略受到越来越多的关注.7-9
糖酵解途径是依赖细胞膜上的葡萄糖转运体将胞外的葡萄糖转运进胞内,通过己糖激酶(HK)、丙酮酸激酶(PK)、乳酸脱氢酶(LDH)、磷酸果糖激酶(PFK)和磷酸甘油醛脱氢酶(GAPDH)等糖酵解酶分解代谢,生成终产物丙酮酸.10研究表明这些糖酵解酶在恶性肿瘤中高表达,它们均有可能成为通过糖酵解途径靶向治疗恶性肿瘤的靶点.11丙酮酸激酶(PK)是糖酵解途径中最重要的限速酶之一,在哺乳动物中,有2种不同的基因及产物,人体的大部分组织表达为丙酮酸激酶M(PKM)中的2种亚型PKM1或PKM2,所有的肿瘤细胞都表达PKM2,而分化组织多表达PKM1.12PKM2在低活性态和高活性态之间存在平衡,PKM2的活性形式主要以四聚体存在.据文献13报道,在肿瘤细胞中,PKM2主要以低活性形式存在,与此相反,PKM1在其原生状态具有较高的活性.在功能上,肿瘤细胞中PKM2的活性下调被认为有助于调节关键糖酵解途径的朝向,有利于肿瘤细胞利用糖酵解途径的中间产物作为其合成脂质、氨基酸、核酸等的前体.14综上所述,激活PKM2的治疗策略可能会将肿瘤细胞纠正为正常的细胞代谢水平,并恢复正常分化细胞的状态特性.研究者们基于该思路已经设计和合成了多种PKM2激动剂,如双磺酰胺类、磺酰吲哚类、噻吩并吡咯并吡嗪酮类等,这些小分子药物中研发进度最快的已经完成了II期临床试验.15-19随着PKM2多聚体晶体结构的阐明,尤其是双磺酰胺类、磺酰吲哚类和噻吩并吡咯并吡嗪酮类激动剂与PKM2四聚体复合物的晶体结构被解析出来,以及计算机辅助药物设计方法的应用,为基于靶点结构的PKM2激动剂设计和研发提供了可能.
近年来,文献20,21报道了小分子PKM2抑制剂的合成和活性研究,在体外和体内抗肿瘤试验中均表现出了良好的药效.但小分子化合物和PKM2的定量构效关系(QSAR)研究尚未见文献报道.本文采用基于多复合物的药效团模型探讨了芳基磺酰胺类化合物与PKM2的相互作用模式,并利用CoMFA和CoMSIA方法对一组芳基磺酰胺类衍生物类PKM2激动剂进行3D-QSAR研究,22,23讨论了其三维静电场、立体场空间结构与抗肿瘤活性之间的关系,以期得到较好的QSAR模型;分析了结构与活性的关系,本文的研究结果将为进一步设计、合成新型的PKM2激动剂提供理论基础.
2 实验部分
2.1 化合物的选择
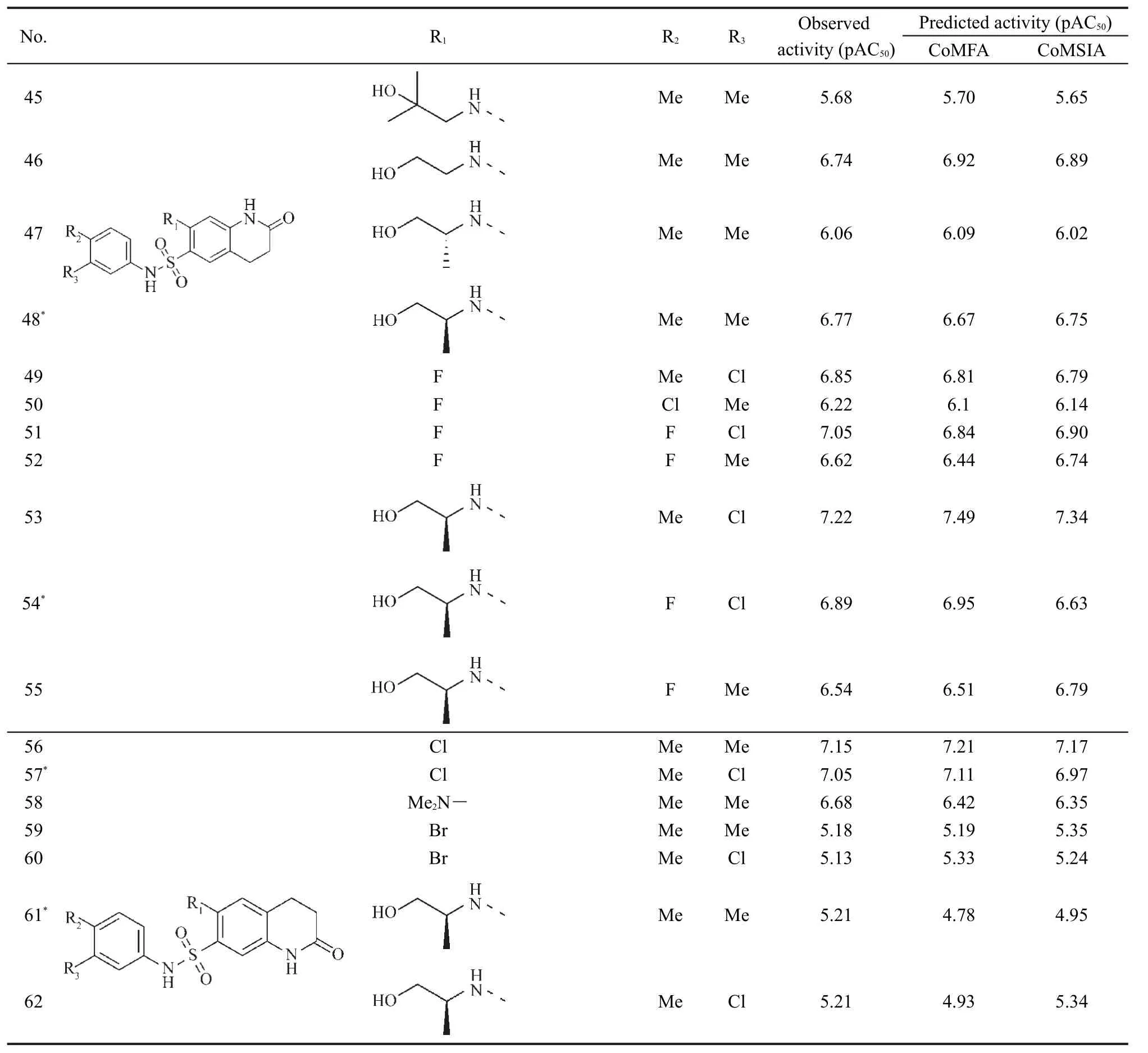
选取结构上具有相同骨架的62个具有类似骨架的芳基磺酰胺类PKM2激动剂为研究对象,20,21其结构如表1所示,它们均由同一个研究小组合成并测定活性的,这些化合物对PKM2的激动活性数据测定条件、方法相同,数据具有可比性.它们的生物活性指标用半数激动常数AC50的负对数,即pAC50进行量度,化合物的活性值列于表l.在具体计算时,随机选取13个化合物(表中带*号的化合物)作为测试集来评价模型的预测能力,其余49个化合物作为训练集产生3D-QSAR模型.本研究所有工作都是在Sybyl-X 2.0软件上完成的,计算中如未经特别指明,各参数均为Sybyl默认值.24
2.2 建立基于多复合物的药效团模型
用于生成药效团模型的6个PKM2-激动剂复合物晶体结构从蛋白质结构数据库(PDB)获得,25各复合物的详细信息见表2.首先将所有晶体结构使用Discovery Studio(DS)软件的“Multiple Structure Alignment”模块叠合到一起,26叠合方式为蛋白质主链的α碳原子叠合,其他选项均为默认设置.使用Discovery Studio软件的“Receptor-Ligand Pharmacophore Generation”模块对每个叠合后的复合物分别生成药效团,所有的药效团特征与PKM2蛋白结构叠合见图1,表2统计分析了所有的药效团特征,具有代表性的共同药效团模型如图2所示.
2.3 活性构象确定及分子叠合

表1 PKM2激动剂的结构与活性Table 1 Structures and activities of PKM2 activators
continued Table 1
在CoMFA和CoMSIA计算中的关键步骤之一是确定所研究分子的活性构象,并对这些活性构象进行合理的叠合,叠合结果的好坏将直接影响最终模型的好坏,是CoMFA和CoMSIA计算能否成功的前提条件.27本文利用基于多复合物生成的共同药效团模型产生的分子构象作为其活性构象,采用基于药效团特征的叠合方式,以化合物中活性最高的分子53为模板,Discovery Studio软件的“Ligand Pharmacophore Mapping”模块将其余训练集化合物与模板分子进行骨架叠合.
2.4CoMFA模型的建立
CoMFA分析在Sybyl-X 2.0软件包的QSAR模块中进行.分析力场包括静电场和立体场,本实验考察步长(0.05-0.2 nm)以及静电场能和立体场能的截断值(energy cutoff:4.187-502.4 kJ·mol-1)对CoMFA结果的影响.以sp3杂化的C+为探针,采用偏最小二乘法(PLS)对分子周围分布的势场值和生物活性进行回归分析.28-30实验中先用留一(LOO)法进行交叉验证分析,来确定模型的交叉验证相关系数(q2)和最佳主成分数(N).31然后再根据得到的最佳主成分数进行非交叉验证来建立最终的比较分子场模型.
2.5 CoMSIA模型的建立
CoMSIA模型的建立方法与CoMFA类似,在建模的过程中,选取静电场、立体场、疏水场、氢键受体场和给体场考察化合物与受体的相互作用,通过考察不同场的组合寻找具有最好交叉验证系数的分子场.利用所得最佳场组合,进一步研究网格步长(0.05-0.2 nm)和衰减因子(0.2-0.4)对CoMSIA结果的影响.采用LOO法进行交叉验证,得到对应的交叉验证相关系数和最佳主成分数,随后通过非交叉验证进行回归计算,从而建立相应的CoMSIA模型.
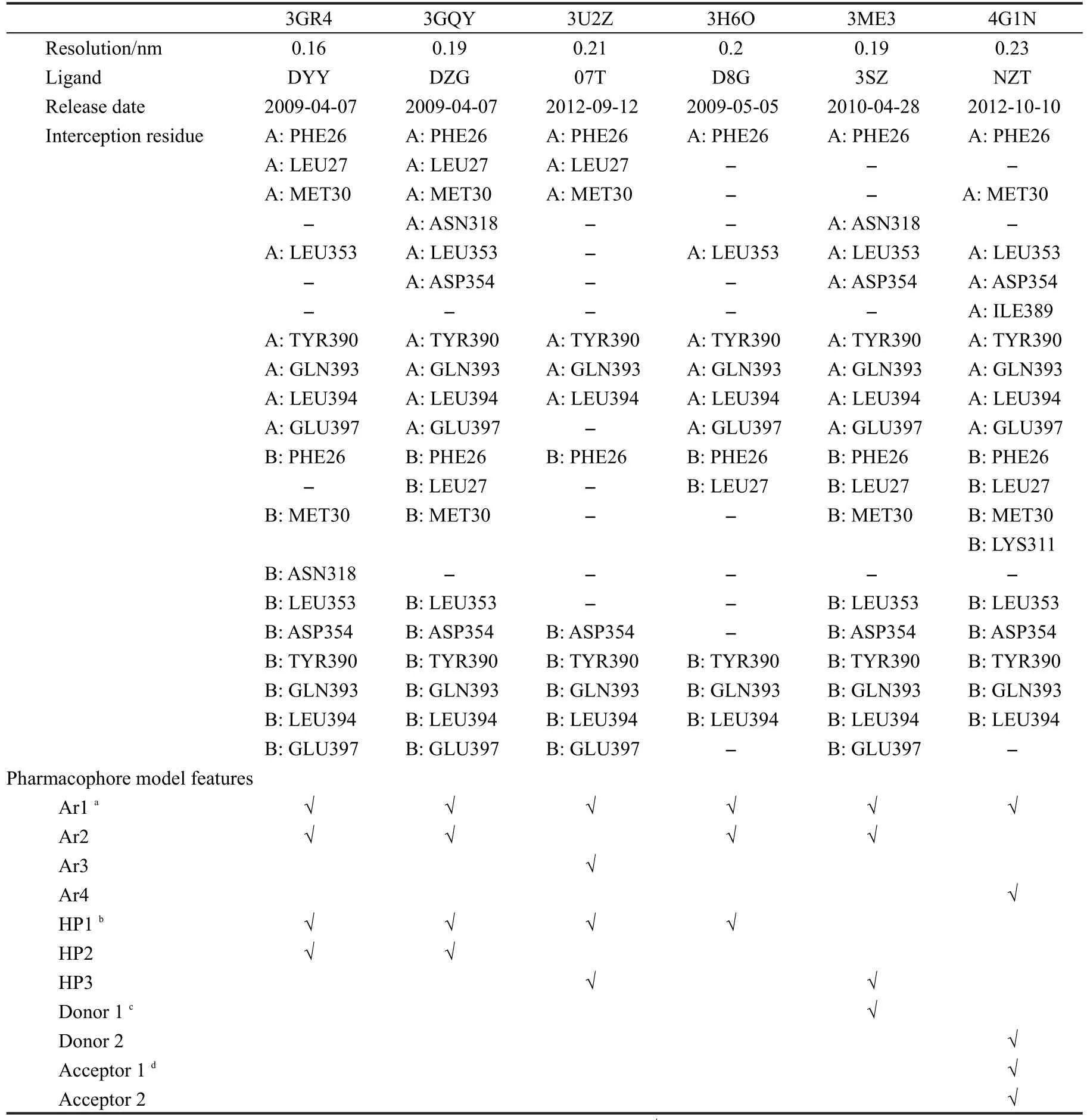
表2 PDB数据库中6个PKM2-激动剂复合物的关键氨基酸残基和药效团特征分析Table 2 Analyses of critical amino acid residues and pharmacophore features for PKM2-activator complexes deposited in protein databank database
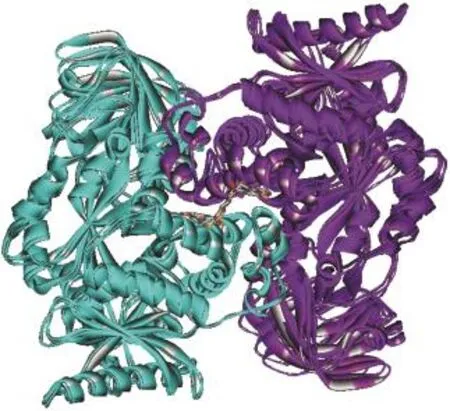
图1 6个PKM2-激动剂复合物叠合示意图Fig.1 Superimposition of six PKM2-activator complexes
2.6 模型验证
QSAR模型采用LOO法进行内部验证,通过未参与建模的测试集分子进行外部验证.
3 结果与讨论
3.1 基于多复合物的药效团模型
用于生成药效团模型的6个PKM2-激动剂复合物晶体结构详细信息见表2.首先以分辨率最高的晶体结构3GR4为模版,将6个晶体结构叠合到一起,晶体结构叠合示意图见图1.由于PKM2与激动剂结合后以四聚体的形式存在,每2个亚基结合1个小分子激动剂,并且A/B亚基与C/D亚基完全对称,为了清楚地显示小分子与大分子间的相互作用,本文所有相互作用的结果均以A/B亚基为研究对象,并且经过比对,C/D亚基与小分子的结合方式以及小分子激动剂构象与A/B亚基完全相同.由图1可见,所有6个PKM2蛋白的A/B亚基都很好地叠合到一起,并且所有的小分子激动剂都位于同一结合位点,说明这些小分子激动剂应该是以类似的方式与PKM2蛋白发生相互作用.所有基于复合物结构产生的药效团特征见表2和图2所示,按性质和与结合位点残基的相互作用可分为11个药效团特征,其中4个芳香环(Ar1-Ar4)、3个疏水相互作用位点(HP1-HP3)、2个氢键供体(D1,D3)和2个氢键受体(A1,A2).在这11个药效团特征中,Ar1、Ar2和HP1这3个药效团特征在大部分的复合物晶体结构中均有出现,说明其应该是小分子激动剂与PKM2具有共通性的相互作用;而其他药效团特征仅在一两个复合物中出现,说明其不具有普遍性,因此选择Ar1、Ar2和HP1作为基于多复合物的共同药效团进行下一步的研究.
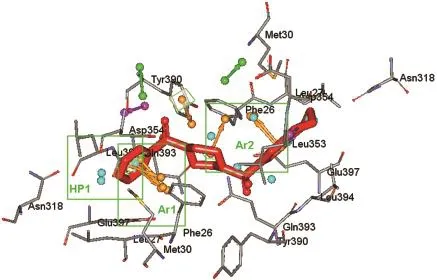
图2 PKM2-激动剂复合物产生的药效团特征Fig.2 Pharmacophore features generated by the PKM2-activator complexes
根据文献报道和课题组的前期研究结果,32-36基于多复合物的共同药效团(MCBP)由于同时考虑了小分子与大分子的相互作用和小分子配体的化学结构特征,可能较单纯基于小分子配体产生的药效团更为全面;而与分子对接相比,MCBP同时考虑了多个复合物中大分子与小分子相互作用的共通特征,分子对接主要考虑的是单个大分子与小分子的相互作用;当应用于虚拟筛选时,MCBP有比分子对接更高的效率,当应用于产生3D-QSAR的活性构象和分子叠合时,基于MCBP的分子叠合的3DQSAR模型与基于分子对接的相当或略好.34为进一步验证MCBP是否能有效地找到小分子化合物的活性构象,选择与产生MCBP的6个复合物中小分子配体的化学结构类型不同的化合物1,使用Discovery Studio软件的“Ligand Pharmacophore Mapping”模块产生构象,拟合方式选择‘flexible’,其他选项均为默认值,选择产生出的小分子构象中与MCBP拟合度最高的与MCBP进行比较,结果如图3所示,化合物1很好地再现了MCBP的药效团特征,两个芳香环分别位于Ar1和Ar2上面,而四氢喹啉环位于HP1附近,说明基于MCBP产生的小分子构象较好地反映了其与PKM2蛋白的作用方式,基于MCBP的小分子化合物构象叠合(见图4)可以用于后续的3D-QSAR研究.
3.2CoMFA和CoMSIA模型
在CoMFA研究中发现,网格点步长选取0.1 nm,静电场能和立体场能的energy cutoff值设为141.1 kJ·mol-1时得到CoMFA模型的结果最为理想.最佳CoMFA模型的统计学参数如表2所示,其中交叉验证相关系数q2=0.545,最佳主成分数为4.一般认为,q2大于0.5时,所得模型具有可靠的预测能力.37由最佳主成分数进行非交叉验证建立的CoMFA模型的非交叉验证相关系数r2=0.966,标准偏差(SEE)为0.146,立体场和静电场的贡献值分别为47.8%和52.2%,表明基团的空间效应和电性分布对活性均有一定影响,静电场的影响高于立体场.
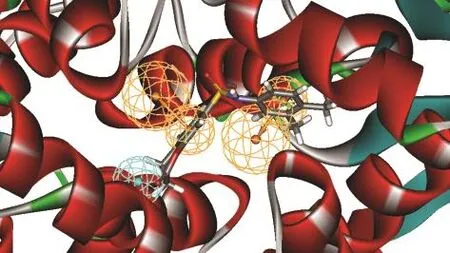
图3 基于多复合物的PKM2激动剂共同药效团特征Fig.3 Multi-complex based common pharmacophore features of PKM2 activators
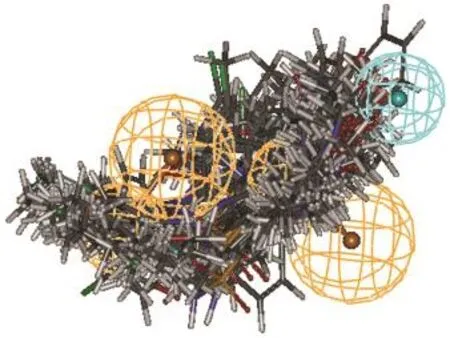
图4 基于MCBP的分子构象叠合图Fig.4 Molecular alignments based on the MCBP
对于CoMSIA模型,选取静电场、立体场、疏水场、氢键受体场和氢键给体场5种场的不同组合来考察化合物与受体的相互作用,发现静电场、立体场和疏水场组合所得结果最佳,当网格点步长为0.1 nm时所得结果最为理想.在CoMSIA计算中,采用高斯函数计算分子的相似性指数,函数中的α为衰减因子,其取值对计算结果有一定影响,最佳值一般在0.2-0.4之间.衰减因子在0.2-0.4范围内CoMSIA模型均得到了理想的统计结果,当衰减因子为0.3时,得到最佳的CoMSIA模型.其统计学参数见表3.
3.3 3D-QSAR模型的验证
为验证所得到的CoMFA及CoMSIA模型的稳定性和预测能力,对训练集中的49个化合物进行活性预测,并将未参加构建模型的测试集的13个结构类化合物放入已建立的3D-QSAR模型中,得到测试集化合物及训练集化合物活性的预测值.CoMFA及CoMSIA模型对62个化合物抑制活性实验值与预测值均列在表1中.将预测值与实验值进行线性拟合,如图5所示.其中测试集分子的实验活性值和CoMFA、CoMSIA预测值之间的相关系数分别为0.966和0.987.从图5可以看出,所有训练集化合物的预测活性和实验活性数据线性关系比较理想,表明由训练集所得的CoMFA、CoMSIA模型都具有较好的相关性.测试集的13个分子的预测值和实验值也都集中在直线附近,实测值与预测值之间的残差不大,这也进一步证明此模型具有较好的预测能力,可以用于预测与骨架结构相似化合物的生物活性.
3.4 三维等值线图
图6是CoMFA立体场和静电场的三维等值线图.黄色区域表示相应位置减小基团的体积有利于提高活性,绿色区域表示此处增加基团的体积有利于提高活性;蓝色区域表示增加化合物的正电性将有利于化合物活性的提高,红色区域表示增加化合物的负电性有利于化合物活性的提高.图中加入的化合物为模板分子53.

表3 最佳CoMFA和CoMSIA模型统计参数Table 3 Statistical parameters of the best CoMFAand CoMSIAmodels
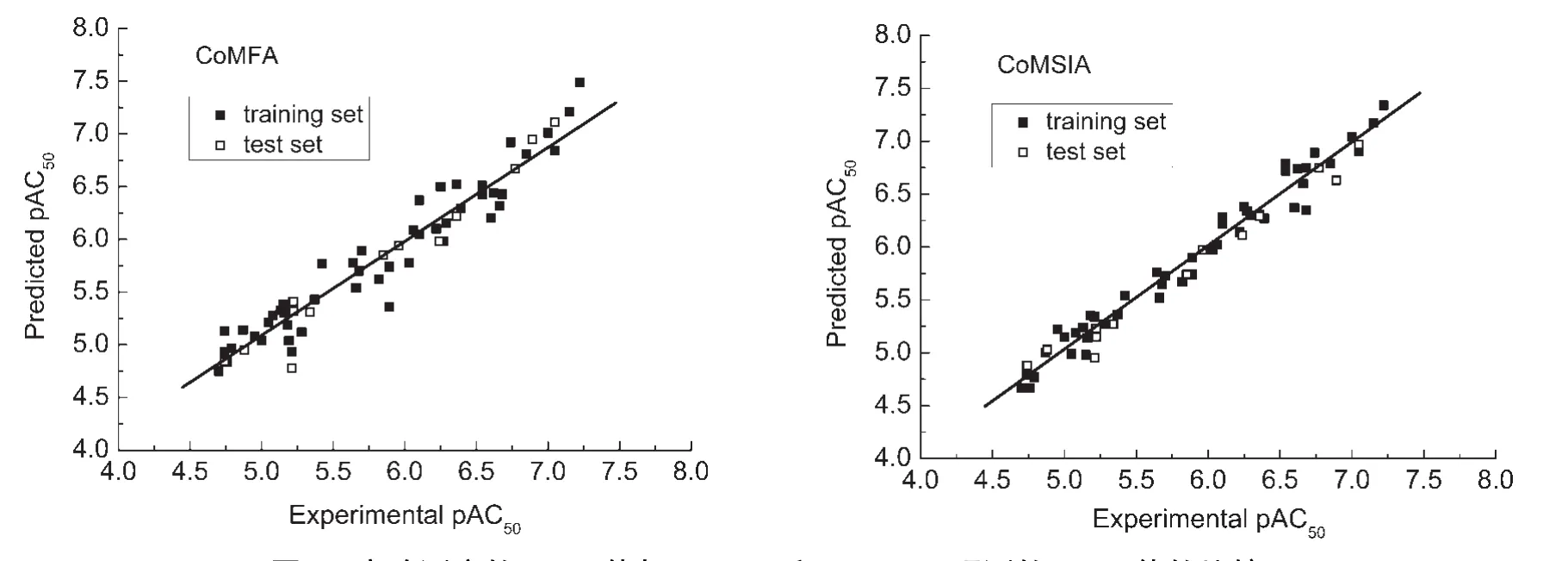
图5 实验测定的pAC50值与CoMFA和CoMSIA预测的pAC50值的比较Fig.5 Comparison of experimental and predicted pAC50for CoMFAand CoMSIA
由图6a可见,在化合物53的R1的右方位置有一块绿色区域,左方有三块较小的黄色区域,说明此处若取代基向右方适当增大体积可以提高化合物的活性,而向其他方向伸展则会降低活性.例如化合物48、53-55,当R1位置为1-羟基-异丙胺基时,化合物的活性相对较高,而当R1位置为其他体积较小基团,例如甲胺基或二甲氨基等取代时,化合物的活性则有不同程度的下降.图中的化合物53的磺酰胺基和R2、R3取代基附近有较大的黄色区域,说明此处不宜有较大的取代基.由图6b可见,在R1所在位右侧有一块蓝色区域,说明在此处若连接有吸电子基团时,可以降低电子云密度,将有利于提高活性,例如化合物38-62在R1位置上连有卤素或含氮基团,其活性普遍要比无取代基的化合物活性高;从图中还可看出,在四氢喹啉取代基的2位有一块红色区域,说明此处若含带负电基团可增加化合物活性;在R2取代基所在位置附近有一块红色区域,故R2取代基带有吸电子基团可以增加化合物的活性.模型中立体场的贡献(47.8%)与静电场的贡献(52.2%)相当,所以对化合物的修饰改造应均衡考虑空间位阻和静电效应的要求.
图7为CoMSIA模型的立体场、静电场、疏水场和氢键受体场等值线图.在图7a中,绿色区域表示引入大体积取代基将有利于化合物活性的提高,黄色区域表示引入大体积取代基将有可能降低化合物的活性.图7b的蓝色区域表示增加化合物的正电性将有利于化合物活性的提高,红色区域表示增加化合物的负电性有利于化合物活性的提高.CoMSIA模型的立体场和静电场分布趋势均主要在R1取代基所在位置,表明R1取代基应控制好取代基的伸展方向,并且带负电的吸电子取代基应处于末端位置.在疏水场等值线图中,黄色区域表示增加疏水性有利于提高活性,白色区域表示减少疏水性有利于化合物活性的提高,如图7c所示,在R1、R2和R3所在位置均有黄色区域,表明在这些位置提高取代基疏水性有利于增加活性.
3.5 基于3D-QSAR的分子设计
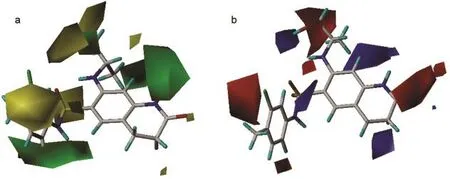
图6 CoMFA模型三维等值线图Fig.6 Three dimensional(3D)contour plots of the CoMFAmodel
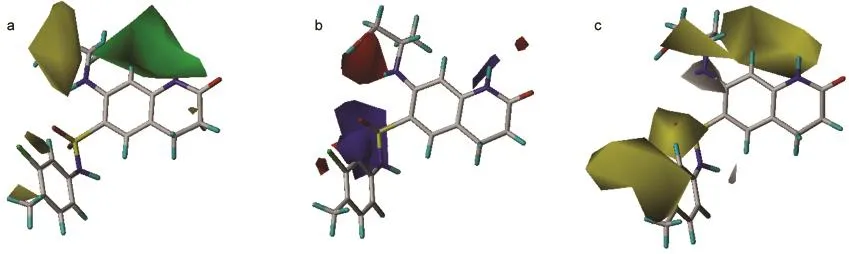
图7 CoMSIA模型的三维等值线图Fig.7 3D contour plot of the CoMSIAmodel

表4 设计化合物的预测活性Table 4 Predicted activities of designed compounds

图8 设计化合物的结构Fig.8 Structures of designed compounds
QSAR研究的目的是根据模型设计新化合物并运用该模型预测新化合物的活性,为先导化合物的设计合成提供理论依据.38根据前面已建立的3DQSAR模型及有关分析结果,现以表1中生物活性较高的化合物为起始化合物,结合药效团模型和课题组的研究基础,尝试对其分子结构进行修饰,新设计的化合物结构如图8所示,用建立的3D-QSAR模型进行活性预测,得到的CoMFA、CoMSIA预测值见表4.利用R3的苯环或咪唑环以及四氢喹啉环满足药效团模型的要求,同时四氢喹啉环的3,4位用一个四氢呋喃环控制空间取向,同时也规避了大部分相关专利的保护;R4取代基引入二甲氨基或吗啉环以满足此处空间位阻和静电需求,同时可以增加整个分子的水溶性;该系列化合物的生物活性有待进一步实验的验证.
4 结论
综合采用基于多复合物的药效团模型、CoMFA和CoMSIA方法建立了3D-QSAR模型,交叉验证的相关系数分别为0.545和0.653,表明该模型具有良好的预测能力,其三维等值线图直观地解释了化合物的定量构效关系,从立体场、静电场和疏水场等方面全面地揭示了影响化合物活性的分子结构特征.对于设计新结构的PKM2激动剂来说,在保证化合物合适的空间结构取向以满足药效团模型的基础上,在四氢喹啉环的2位宜引入带吸电子能力基团;6,7位应注意控制基团的大小,避免引入较大基团;5,8位取代基则可适当增大基团的体积.这些结论为进一步结构优化、设计和合成新型的PKM2激动剂提供了理论依据.
(1)Tennant,D.A.;Duran,R.V.;Gottlieb,E.Nature Rev.Cancer 2010,10,267.doi:10.1038/nrc2817
(2)Vander Heiden,M.G.Nature Rev.Drug Discov.2011,10,671.doi:10.1038/nrd3504
(3) Cairns,R.A.;Harris,I.S.;Mak,T.W.Nature Rev.Cancer 2011,11,85.
(4) Levine,A.J.;Puzio-Kuter,A.M.Science 2010,330,1340.doi:10.1126/science.1193494
(5) Warburg,O.Science 1956,123,309.doi:10.1126/science.123.3191.309
(6)Vander Heiden,M.G.;Cantley,L.C.;Thompson,C.B.Science 2009,324,1029.doi:10.1126/science.1160809
(7) Dang,C.V.;Kim,J.W.;Gao,P.;Yustein,J.Nat.Rev.Cancer 2008,8,51.doi:10.1038/nrc2274
(8) Robey,R.B.;Hay,N.Semin.Cancer Biol.2009,19,25.doi:10.1016/j.semcancer.2008.11.010
(9) Shaw,R.J.Curr.Opin.Cell Biol.2006,18,598.doi:10.1016/j.ceb.2006.10.005
(10) Diaz-Ruiz,R.;Rigoulet,M.;Devin,A.BiochimicaetBiophysica Acta-Bioenergetics 2011,1807,568.doi:10.1016/j.bbabio.2010.08.010
(11)Dombrauckas,J.D.;Santarsiero,B.D.;Mesecar,A.D.Biochemistry 2005,44,9417.doi:10.1021/bi0474923
(12)Takenaka,M.;Yamada,K.;Lu,T.;Kang,R.;Tanaka,T.;Noguchi,T.Eur.J.Biochem.1996,235,366.doi:10.1111/ejb.1996.235.issue-1-2
(13)Christofk,H.R.;Vander Heiden,M.G.;Harris,M.H.;Ramanathan,A.;Gerszten,R.E.;Wei,R.;Fleming,M.D.;Schreiber,S.L.;Cantley,L.C.Nature 2008,452,230.doi:10.1038/nature06734
(14)Mao,J.Z.;Guo,W.H.;Shi,H.S.;Zhao,Y.W.;Yang,H.S.Journal of Sichuan University(Medical Science Edition)2011,42,308.[毛婧芝,郭文浩,石华山,赵玉伟,杨寒朔.四川大学学报(医学版),2011,42,308.]
(15) Boxer,M.B.;Jiang,J.K.;Vander Heiden,M.G.;Shen,M.;Skoumbourdis,A.P.;Southall,N.;Veith,H.;Leister,W.;Austin,C.P.;Park,H.W.;Inglese,J.;Cantley,L.C.;Auld,D.S.;Thomas,C.J.J.Med.Chem.2010,53,1048.doi:10.1021/jm901577g
(16)Thomas,C.J.;Boxer,M.B.;Jiang,J.K.;Vander Heiden,M.G.;Walsh,M.J.;Brimacombe,K.;Shen,M.;Skoumbourdis,A.P.;Park,H.W.;Cantley,L.C.;Auld,D.S.Abstr.Pap.Am.Chem.Soc.2011,242,233-MEDI.
(17)Anastasiou,D.;Poulogiannis,G.;Asara,J.M.;Boxer,M.B.;Jiang,J.K.;Shen,M.;Bellinger,G.;Sasaki,A.T.;Locasale,J.W.;Auld,D.S.;Thomas,C.J.;Vander Heiden,M.G.;Cantley,L.C.Science 2011,334,1278.doi:10.1126/science.1211485
(18)Anastasiou,D.;Yu,Y.M.;Israelsen,W.J.;Jiang,J.K.;Boxer,M.B.;Hong,B.S.;Tempel,W.;Dimov,S.;Shen,M.;Jha,A.;Yang,H.;Mattaini,K.R.;Metallo,C.M.;Fiske,B.P.;Courtney,K.D.;Malstrom,S.;Khan,T.M.;Kung,C.;Skoumbourdis,A.P.;Veith,H.;Southall,N.;Walsh,M.J.;Brimacombe,K.R.;Leister,W.;Lunt,S.Y.;Johnson,Z.R.;Yen,K.E.;Kunii,K.;Davidson,S.M.;Christofk,H.R.;Austin,C.P.;Inglese,J.;Harris,M.H.;Asara,J.M.;Stephanopoulos,G.;Salituro,F.G.;Jin,S.F.;Dang,L.;Auld,D.S.;Park,H.W.;Cantley,L.C.;Thomas,C.J.;Heiden,M.G.V.Nat.Chem.Biol.2012,8,839.doi:10.1038/nchembio.1060
(19)Anastasiou,D.;Yu,Y.M.;Israelsen,W.J.;Jiang,J.K.;Boxer,M.B.;Hong,B.S.;Tempel,W.;Dimov,S.;Shen,M.;Jha,A.;Yang,H.;Mattaini,K.R.;Metallo,C.M.;Fiske,B.P.;Courtney,K.D.;Malstrom,S.;Khan,T.M.;Kung,C.;Skoumbourdis,A.P.;Veith,H.;Southall,N.;Walsh,M.J.;Brimacombe,K.R.;Leister,W.;Lunt,S.Y.;Johnson,Z.R.;Yen,K.E.;Kunii,K.;Davidson,S.M.;Christofk,H.R.;Austin,C.P.;Inglese,J.;Harris,M.H.;Asara,J.M.;Stephanopoulos,G.;Salituro,F.G.;Jin,S.F.;Dang,L.N.;Auld,D.S.;Park,H.W.;Cantley,L.C.;Thomas,C.J.;VanderHeiden,M.G.Nat.Chem.Biol.2012,8,1008.
(20) Jiang,J.K.;Boxer,M.B.;Vander Heiden,M.G.;Shen,M.;Skoumbourdis,A.P.;Southall,N.;Veith,H.;Leister,W.;Austin,C.P.;Park,H.W.;Inglese,J.;Cantley,L.C.;Auld,D.S.;Thomas,C.J.Bioorganic&Medicinal Chemistry Letters 2010,20,3387.doi:10.1016/j.bmcl.2010.04.015
(21)Walsh,M.J.;Brimacombe,K.R.;Veith,H.;Bougie,J.M.;Daniel,T.;Leister,W.;Cantley,L.C.;Israelsen,W.J.;Vander Heiden,M.G.;Shen,M.;Auld,D.S.;Thomas,C.J.;Boxer,M.B.Bioorganic&Medicinal Chemistry Letters 2011,21,6322.doi:10.1016/j.bmcl.2011.08.114
(22) Kubinyi,H.Drug Discov.Today 1997,11,457.
(23) Kubinyi,H.Drug Discov.Today 1997,12,538.
(24) SYBYL Molecular Modeling Software;Tripos Inc.:St.Louis,MO,USA,2012.
(25) http://www.rcsb.org
(26) Discovery Studio,version 3.5;Accelrys Inc.:San Diego,CA,USA,2012.
(27)Kang,C.M.;Zhao,X.H.;Wang,X.Y.;Cheng,J.G.;Lü,Y.T.Acta Phys.-Chim.Sin.2013,29,431.[康从民,赵绪浩,王新宇,程家高,吕英涛.物理化学学报,2013,29,431.]doi:10.3866/PKU.WHXB201211151
(28)McIntosh,A.R.;Bookstein,F.L.;Haxby,J.V.;Grady,C.L.Neuroimage 1996,3,143.doi:10.1006/nimg.1996.0016
(29) Cramer,R.D.Perspect Drug Discov.Des.1993,1,269.doi:10.1007/BF02174528
(30) Qiao,K.;Zeng,L.X.;Jin,H.W.;Liu,Z.M.;Zhang,L.R.Acta Phys.-Chim.Sin.2012,28,1509.[乔 康,曾凌晓,金宏威,刘振明,张亮仁.物理化学学报,2012,28,1509.]doi:10.3866/PKU.WHXB201203272
(31) Tropsha,A.;Golbraikh,A.J.Mol.Graph.Model.2002,20,269.doi:10.1016/S1093-3263(01)00123-1
(32) Chaudhaery,S.S.;Roy,K.K.;Saxena,A.K.J.Chem Inf.Model.2009,49,1590.doi:10.1021/ci900049e
(33)Ouyang,L.;He,G.;Huang,W.;Song,X.R.;Wu,F.B.;Xiang,M.L.Int.J.Mol.Sci.2012,13,5348.doi:10.3390/ijms13055348
(34)He,G.;Qiu,M.H.;Li,R.;Ouyang,L.;Wu,F.B.;Song,X.R.;Cheng,L.;Xiang,M.L.;Yu,L.T.Chem.Biol.Drug Des.2012,79,960.doi:10.1111/jpp.2012.79.issue-6
(35) He,G.;Qiu,M.H.;Li,R.;Song,X.R.;Zheng,X.;Shi,J.Y.;Xu,G.B.;Han,J.;Yu,L.T.;Yang,S.Y.;Chen,L.J.;Wei,Y.Q.Molecular Simulation 2011,37,31.doi:10.1080/08927022.2010.517529
(36) Wu,F.B.;Xu,T.;He,G.;Ouyang,L.;Han,B.;Peng,C.;Song,X.R.;Xiang,M.L.Int.J.Mol.Sci.2012,13,15668.doi:10.3390/ijms131215668
(37) Clark,M.;Cramer,R.D.;Jones,D.M.;Patterson,D.E.;Simeroth,P.E.Tetrahedron Comput.Method 1990,3,47.doi:10.1016/0898-5529(90)90120-W
(38)Dai,Z.J.;Zhou,W.;Yuan,Z.M.Acta Phys.-Chim.Sin.2011,27,1654.[代志军,周 玮,袁哲明.物理化学学报,2011,27,1654.]doi:10.3866/PKU.WHXB20110735
